Jama Software is always looking for news that would benefit and inform our industry partners. As such, we’ve curated a series of customer and industry spotlight articles that we found insightful. In this blog post, we share an article, sourced from EURACTIVE, titled “The AI Act’s Fine Line On Critical Infrastructure” – originally authored by Luca Bertuzzi on February 8, 2023.
The AI Act’s Fine Line On Critical Infrastructure
As EU policymakers make progress in defining an upcoming rulebook for Artificial Intelligence, the question of to what extent AI models employed to manage critical infrastructure should be covered by tight requirements still remains open.
The AI Act is reaching a critical stage in the legislative process, with the European Parliament set to reach a common position in the coming weeks. The legislative proposal is the world’s first attempt to put in place a comprehensive set of rules for Artificial Intelligence based on its potential risks.
A critical aspect of the draft law is the category of AI models that can cause significant harm, which must comply with stricter obligations regarding quality and risk management. However, concerning critical infrastructure, how to assess risk remains a matter of debate.
AI in critical infrastructure
Artificial Intelligence is increasingly employed in managing critical infrastructure, notably for project development, maintenance and performance optimization.
An example on the construction side is Sweco Netherlands, an engineering consultancy company tasked to extend the light-rail system of Bybanen, Norway’s second-largest city, considering the existing tram lines, adjacent roads, cycle lanes, pedestrian zones and surrounding public areas.
To put together these different factors, Sweco NL used a digital twin model to visualise its project and understand how design changes would impact the timeline, costs and surroundings. The company estimates it reduced construction errors by 25% as a result.
Another area of application for this technology is dams. In 2017 HDR, a US construction company, applied machine learning to a dam’s digital twin model to simulate how the infrastructure would be affected by changes like natural shifting and erosion of the surrounding soil over time.
The model allowed dam operators to detect anomalies like cracks with an accuracy of two centimeters, differentiating them from harmless algae growth, and taking corrective measures before they grew into more significant problems.
The original AI Act proposal noted that “it is appropriate to classify as high-risk the AI systems intended to be used as safety components in the management and operation of road traffic and the supply of water, gas, heating and electricity.”
In the EU Council of Ministers, member states clarified that the concept of the safety component should be distinguished by the management system itself. In other words, in a dam, the mechanism to open the volts is the management system, whilst the technology that monitors the water pressure is a safety component.
In the European Parliament, the MEPs spearheading the work on the AI Act proposed to differentiate the management of traffic on roads, rail and air, from supply networks like water, gas, heating, energy and electricity, in compromise amendments obtained by EURACTIV.
While the Council included digital infrastructure like cloud services and data centres in the list of high-risk use cases, since the intent is to prevent “appreciable disruptions in the ordinary conduct of social and economic activities”, EU lawmakers have so far not done so.
The addition to the high-risk list caused significant anxiety in the telecom industry, which uses AI to manage network capacity, plan upgrades, detect frauds and improve energy efficiency. The question is whether the malfunction of any of these algorithms might bring the whole system down.
For example, if a telecom operator miscalculates traffic peaks in different areas of its network, would that lead to internet outages? A representative of telecom operators told EURACTIV they are not aware of any situation where that occurred, branding the issue as ‘highly hypothetical’.
More generally, critical infrastructure operators are concerned that, by casting the high-risk category of the AI regulation too wide, they might be precluded from useful tools that contribute to making their systems more efficient and secure.
A case in point is that member states excluded AI-powered cybersecurity tools from the definition of safety component.
Anti-virus malware analysis is based on predictive models and machine learning, meaning critical infrastructure service providers would have been precluded from using virtually all commercially available anti-viruses.
At the same time, AI-powered management systems are not without risks. Kris Shrishak, a technologist at the Irish Council for Civil Liberties, made the case of India in 2012 when a miscalculation of the electric grid’s peak traffic led to perhaps the largest blackout in history.
Therefore, the argument for a more granular approach in the high-risk categorisation relates to when the AI solutions make the infrastructure safer and if their failure does not entail an imminent threat.
Physical maintenance, for instance, is often costly and time-consuming, which might lead to infrastructures falling into disrepair. Not employing AI’s capacity to identify patterns and spot anomalies before they develop into bigger problems can also come at a cost.
Last year, amid the Russian-prompted energy crisis, France, usually Europe’s largest energy exporter, became a net importer as a record number of its nuclear reactors were put out of service due to maintenance stoppages.
Revolutionizing the Supply Chain: Developments in the Warehouse Robotics Industry
Warehouse robotics is witnessing steady growth, driven by the increasing adoption of automated solutions in storage for food and beverages, consumer goods, retail, and third-party logistics. The collaboration between the e-commerce sector and warehouse robotics is also a major driver of this market, as it allows for developing increasingly sophisticated warehouse automation systems. Additionally, the advent of autonomous mobile robots (AMRs) and the rising popularity of automated guided vehicles (AGVs) are also fueling the growth of this market.
Reforming Warehouse Operations: Increasing Adoption of AMRs and AGVs
One of the most notable trends in the warehouse robotics market is the increasing adoption of AMRs and AGVs. These robots are allowing for the development of more efficient and cost-effective warehouse operations, as they can automate tasks such as picking, packing, and transporting goods. Additionally, the use of AMRs and AGVs is also allowing for the development of more flexible and scalable warehouse operations, which can better meet the changing needs of businesses.
Efficient and Cost-effective: Rising Collaboration between eCommerce Sector and Warehouse Robotics
Another major trend in the warehouse robotics market is the increasing collaboration between the e-commerce sector and warehouse robotics. This is being driven by the growing demand for efficient and cost-effective warehouse operations, as e-commerce businesses look to keep pace with the rapid growth in online sales. Additionally, the use of warehouse robotics is also allowing e-commerce businesses to better meet the changing needs of their customers by providing faster and more accurate delivery of goods.
Advanced Automation: Increasing Preference for Advanced Robots in Warehouses
As the warehouse robotics market continues to evolve, it is clear that there will be a growing demand for advanced robots in warehouses. This is being driven by the surge in the adoption of robots during the COVID-19 outbreak, as businesses look to automate their operations and reduce their dependence on human labor. Additionally, the increasing adoption of industrial automation and control solutions is also contributing to the growth of this market.
Global Perspective: North America to Lead, Europe to hold Second Largest Share
The warehouse robotics market is a global industry, and it is clear that different regions are at different stages of development. Currently, North America is the largest market for warehouse robotics, driven by the rapid adoption of warehouse automation and the presence of major players in the market such as the United States. However, Europe is expected to hold the second-largest market share, driven by the rising adoption of industrial automation and control solutions in the region.
Innovations in Warehouse Automation: A Look at Leading Players’ Latest Developments
The warehouse robotics market is a highly competitive industry with several leading players. Some of the major players in the market include KUKA AG, FANUC Corporation, ABB, and Yaskawa Electric Corporation. These companies have made significant investments in the development of warehouse automation solutions and have a strong presence in the market.
Additionally, there are also several startups and smaller companies that are emerging in the market, such as Locus Robotics and GreyOrange, which are focusing on specific areas of warehouse automation. The competitive landscape is expected to become more intense with the entry of new players and increasing investments in the development of warehouse automation solutions.
In the warehouse robotics market, major players such as Kuka AG, FANUC Corporation, and ABB Limited have been focusing on developing advanced robots for warehouse automation. Recently, Kuka AG announced the launch of its new autonomous mobile robot, LBR iiwa, which is designed for use in the logistics and manufacturing sectors. Similarly, FANUC Corporation has introduced its new autonomous mobile robot, CRX-10iA, which is designed for use in the logistics and distribution sectors.
ABB Limited has also recently launched its new autonomous mobile robot, YuMi, which is designed for use in small parts assembly and other applications in the manufacturing sector. These advancements by leading players demonstrate the increasing focus on developing advanced warehouse automation solutions in the market.
In this blog, we’ll recap our whitepaper, “Understanding Integrated Risk Management for Medical Devices” – To read the entire paper, click HERE.
Understanding Integrated Risk Management for Medical Devices
Knowledge on best practices, how to integrate risk-based thinking into product development cycles, and the importance of having end-to-end traceability to improve risk management, shared by industry and solution experts.
A level of risk exists with all medical devices, no matter how simple they are.Companies developing medical devices are constantly considering who (or what environment, facility, etc.) could potentially be hurt by a device so they can help reduce risk and meet regulatory requirements. Risk management in the context of ISO 14971 is designed to support medical device manufacturers with these tasks — but not all approaches are equal.
The amount of time it takes to manage risks, connect specific risks to specific requirement tasks, and pull together required documents to respond to an audit varies slightly depending on the approach. The risk management process is an integrated process that not only includes teams in product development, quality, but also many other parts of an organization.
This whitepaper taps into the knowledge of industry and solution experts to uncover best practices, how to integrate risk-based thinking into product development cycles, and the importance of having end-to end traceability to improve risk management. Before we dig into integrated risk management, let’s first define some key terms.
Harm – Harm occurs when people are injured physically or their health is compromised or when property or the environment is damaged.
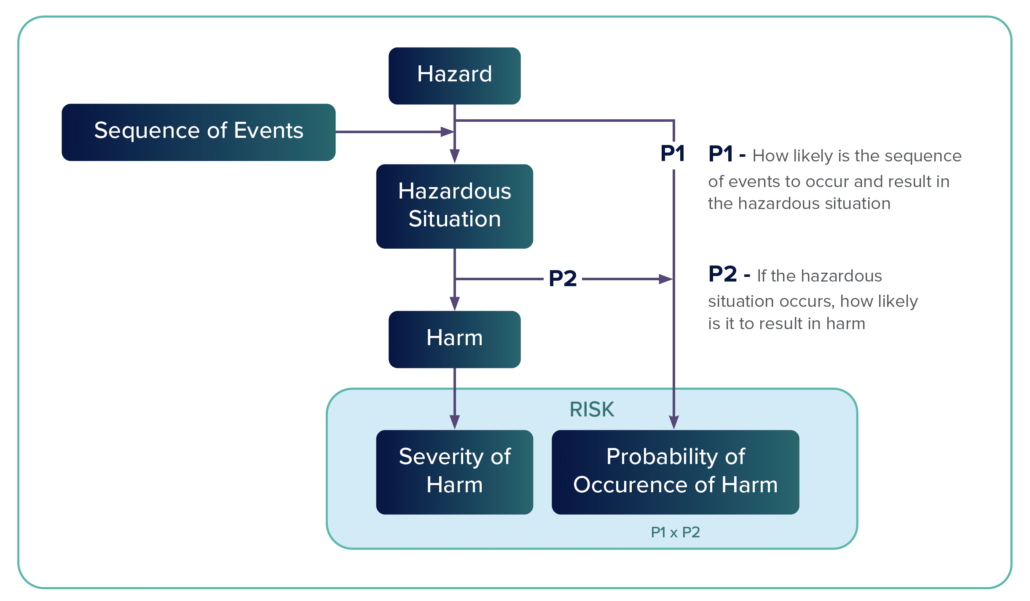
Hazard – A hazard is a potential source of harm. Annex E.2 categorizes hazards in the following way: energy hazards, chemical hazards, biological hazards, operationalhazards, and informational hazards.
Hazardous – A hazardous situation occurs when people are exposed to a hazard or when property or the environment is threatened. A hazardous situation exists when a vulnerable entity is exposed to a hazard.
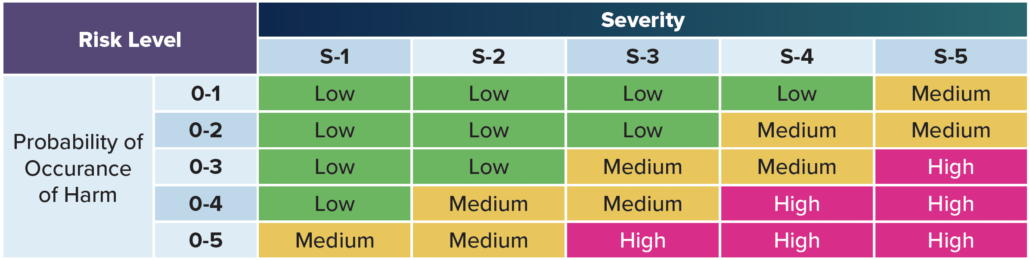
Situation – According to ISO 14971, the concept of risk combines two variables: the probability of harm and the severity of harm.
Risk – For example, if a particular hazardous situation is very likely to cause harm and would be very harmful if it actually occurred, then it would be a high risk situation. Conversely, if it’s very unlikely to cause harm and would be only slightly harmful if it actually occurred, then it would be a trivial risk.
Risk Analysis – Risk analysis is a systematic process that is used to identify hazards and to estimate risk. It includes an examination of every reasonably foreseeable sequence or combination of events that could produce a hazardous situation and cause harm.
Risk Assessment – Risk assessment is a process that is, in turn, made up of two interconnected processes: risk analysis and risk evaluation.
Risk Evaluation – Risk evaluation is a process that is used to examine the estimated risk for each hazardous situation and then to use risk acceptability criteria to determine whether
or not the estimated risk is acceptable and to decide if risk reduction is required.
Risk Control – Risk control is a process that is used to consider risk control options and to select and implement risk control measures that will reduce risk or maintain risk within
specified levels. ISO 14971 expects you to consider the following risk control options and, if possible, to apply them in the following order:
Design safety into the product.
Establish protective measures.
Provide safety information.
Risk Estimation – Risk estimation is a process that is used to assign qualitative or quantitative probability values and severity values to each hazardous situation. These values are then used to estimate risk.
Risk Management – Risk management uses policies, procedures, and practices to systematically analyze, evaluate, control, and monitor risk.
Safety – Safety is freedom from unacceptable risk. Risk acceptability criteria are used to help decide whether or not a risk is unacceptable.
Severity – Severity is a measure of the possible harmful consequences that a hazard could potentially cause.
During risk management — after one defines a device’s intended use(s) — risk analysis can begin with identifying all potential hazards, and hazardous situations. Once this is defined, risk can be estimated and can determine the type of appropriate risk control required. Once the risk controls are implemented, residual risk needs to be analyzed to ensure that the benefits outweigh the risks. Let’s take a look at what’s involved in the risk management process.
Identifying Hazards
“Risk” is defined as the severity and probability that harm will occur. Defining the severity of harm requires you to identify all the known and foreseeable hazards for both intended and unintended uses.
For example, let’s say you have an infusion pump, and that pump has air in the line, which creates a hazardous situation for the patient. Different levels of patient harm can occur, so it’s about uncovering the possible scenarios and the likelihood of a situation’s occurring.
Understanding Harm
Understanding harm includes both people and property. A medical device that catches fire might threaten property, while an infusion pump with air in the line might threaten human life. Think about what could cause harm to people, like a shark swimming in the water. A shark that attacks a person could create different levels of harm. A few examples include loss of a limb, an infection from getting bitten and loss of life. The various levels of harm result from the hazardous situation, which is the shark in the water.
Risk Evaluation
Risk evaluation involves comparing an estimated risk against a specific criterion to determine if a risk is acceptable. Five different levels to evaluate risk are common practice, but you can use as many as you’d like. The most severe risk (level five) might include death or impairment. Level one might include no risk to a patient or operator. The levels inbetween include all the other varying
degrees of risk.
Sequence of Events
A hazardous event includes a number of steps, which is the sequence of events. A risk situation might have two, three, or more steps that, when aligned, create a hazardous event. Risk management tools such as fault trees and failure modes and effects analysis (FMEA) help identify these steps.
Previous version of ISO 14971 used terms like “acceptable” and “unacceptable” to describe risks, but that language has since been removed and the most current version maintains as low as possible (ALAP). The goal of every manufacturer is to lower the risk as much as possible and rethinking how to prioritize risk controls can help.
In this blog, we partially recap this customer story, “Vave Health Migrates to Jama Connect® to Accelerate Development and FDA Clearance” Read the entire story HERE.
Vave Health is committed to revolutionizing the physician-patient experience through innovative, industry-transforming technologies. Their innovative handheld ultrasound device packs the ability to wirelessly connect with your Android or iOS smartphone or tablet.
After initially selecting Matrix Requirements, Vave Health found themselves constrained by the tool’s limited functionality and were ready for a change. Following a requirements management market analysis, Jama Connect® was selected and onboarded due to its ease of use and industry-leading functionality.
Read this customer story to see how now, with more confidence in their processes, Vave health has achieved the following outcomes:
Accelerate the release cadence from what previously took a couple weeks, down to a day or two
Decrease generation of trace matrices from 30 days to one per project
Scale development process with the ability to run multiple projects in parallel
Maintain traceability and instantly identify coverage for verification and validation of requirements to respond to action items sooner in development
VAVE HEALTH CUSTOMER STORY OVERVIEW
CHALLENGES WITH MATRIX
Reports, such as a traceability matrix, were taking too long to generate
The steep learning curve caused most people to revert to working in Word and Excel
Inability to develop parallel projects and reuse data between releases, contributed to duplicated work and slower-than-desired release cadence
SELECTION CRITERIA
A solution that would scale with their growth
Quick-to-adopt and easy-to-use
Strong market presence
Ease of data migration
OUTCOME + FUTURE
Accelerate the release cadence from what previously took a couple of weeks, down to a day or two
Decrease generation of trace matrices from 30 days to one per project
Scale development process with the ability to run multiple projects in parallel
Maintain traceability and instantly identify coverage for verification and validation of requirements to respond to action items sooner in development
In the early days of Vave Health, the development team originally selected Matrix Requirements due to its low cost. While the tool was sufficient for managing their requirements in the preliminary stages of development, as the company began to scale, it became apparent that they needed a more mature, enterprise-grade solution with more robust capabilities.
The main challenges that Vave Health had which led them to seek out a new solution were:
Reports, such as a traceability matrix, were taking too long to generate
Steep learning curve caused most people to revert to working in Word and Excel
Inability to develop parallel projects and reuse data between releases, contributed to duplicated work and slower-than-desired release cadence
As a small team, they did not have dedicated staff to manage requirements – it was a shared responsibility. With Matrix Requirements, the learning curve was so steep, only a few people were able to use it. Even then, it was used similarly to an Excel spreadsheet.
“Matrix Requirements was difficult to use, and it limited our ability to easily extract reports and quickly show traceability. The whole process just took too long,” said Craig Loomis, Vice President of Product at Vave Health.
“One of the deliverables in getting our product released is generating the trace matrix. With Matrix Requirements, it was very cumbersome,” said Sandhya Mitnala, Head of Quality and Regulatory at Vave Health. “We realized that something that should have taken one or two days, and managed through the project, took us almost a month. It was a very manual process.”
Additionally, as the team grew and the development work went from singular to multiple projects, the team ran into limitations using Matrix Requirements.
“One thing we didn’t initially think about when selecting Matrix Requirements was the ability to have multiple projects in motion at the same time,” said Loomis. “Although it was technically possible, there was no good way to extract the trace matrix and manage revisions across different projects at the same time in parallel.”
In order to overcome the limitations of their current tool, the team set out to find a solution that could meet their current needs and grow with them as they expanded in the market.
“The startup world is unique in that you’re trying to do so much more with fewer resources. Sometimes you do need to leverage technology to automate things that larger companies would be
able to throw bodies at,” said Loomis.
When it came time to evaluate the available solutions in the marketplace, things moved quickly.
“From my experience working with Jama Software® at other companies, and my coworkers’ similar experiences, we wanted to move to a more automated solution and Jama Software was on everyone’s mind,” said Mitnala. “It was a very easy choice for us. All of the solutions we looked at, outside of Jama Connect®, were ruled out quickly,” shared Loomis.
During the evaluation process, the Vave Health team was able to access a sandbox account created specifically for them, so they could test out the solution to make sure it was the right fit.
Because there were so many things already in motion, the team wanted to ensure that data migration would not be an issue, so they could keep moving quickly.
“Before we even signed a contract, we spent time in Jama Connect and had a lot of confidence in moving forward. We knew that our data would be migrated easily, and we wouldn’t be putting our projects at risk,” said Loomis.
Although Matrix Requirements supported some initial needs, the team knew that in order to derive the value they needed, it was time to up-level their tool for requirements management and systems engineering. The return on investment for Jama Connect, a robust, yet easy-to-use platform (which comprised the feature set and functionality they required) would increase efficiencies, simplify compliance, reduce risk, and ultimately speed time-to-market, paying dividends in the long run.
“As a startup, the one thing you must ensure is that you are able to move fast. You’re learning the market, you’re working against your competitors, and speed to market is critical. Especially where there are things that can be automated – that’s where you want to invest,” said Loomis.
Jama Software is always looking for news that would benefit and inform our industry partners. As such, we’ve curated a series of customer and industry spotlight articles that we found insightful. In this blog post, we share an article, sourced from Spyrosoft, titled “CE marking for Medical Device Software: a step-by-step guide” – originally authored by MAŁGORZATA KAWAŁKOWSKA.
CE Marking for Medical Device Software: A Step-By-Step Guide
The recent introduction of EU MDR brought many changes to the CE marking certification process for medical devices. There are more requirements to fulfil, which makes the process more complex and much more time-consuming.
To avoid a scenario where the only thing that stops you from releasing your Medical Device Software to the market is the ongoing CE marking certification, you should know when to take the first steps in this process and how to include it in your overall business strategy.
From this blog post, prepared based on an interview with Krzysztof Minicki, Director of Healthcare and Life Sciences, you’ll get to know the steps of the CE marking process for Medical Device Software. You’ll also learn your responsibilities as a manufacturer, and thus get a better understanding of how to plan and optimise the process to avoid possible bottlenecks.
ToC:
Does your software need a CE marking certification?
How to get the CE marking certification for Medical Device Software in the EU?
How to do CE marking self-certification?
How long is the CE marking certification valid?
How much does it cost to get CE marking certification?
How long does the CE marking certification process take?
Need support in the CE marking certification process?
Does your software need a CE marking certification?
Since the introduction of EU MDR in May 2021, the software is now classified as an active medical device. All Medical Device Software that is released to the EU market is required to have the CE marking. Otherwise, it cannot be sold within the EU.
CE MARKING AFTER BREXIT
As of January 1st, 2021, any type of business collaboration between the UK and EU countries got a lot more complicated. So did the CE marking process, which has gone into the transition period ending on January 1st, 2023 – medical companies operating on the British market will be obliged to receive UKCA certification instead of CE marking, with medical products sold in Northern Ireland still requiring the latter. Healthcare products that require to be assessed by a Notified Body will need to be check by an UK Notified Body rather than a EU one.
How to get the CE marking certification for Medical Device Software in the EU?
As a Medical Device Software manufacturer, regardless of whether or not you outsource the manufacturing process, you are responsible for acquiring the CE marking.
In general, the path towards obtaining the CE marking depends on the medical device’s class. Since, in the light of the EU MDR, Medical Device Software is considered as an active medical device, the majority will undergo the upclassification and will belong at least to class IIa.
As per the EU MDR rules, medical devices from classes IIa, IIb and III, have to go through the conformity assessment conducted by a Notified Body. A Notified Body is a third-party organisation, accredited by a European Competent Authority, that checks compliance with the MDR. As a medical device manufacturer, you can select which Notified Body you want to do the assessment for you.
Now, let’s move on now to what the CE marking process for Medical Device Software looks like step by step.
As you determine the classification of your Medical Device Software and appoint a person responsible for regulatory compliance in your organisation, the next step is to create and implement a Quality Management System in accordance with Annex VIII of the MDR and ISO 13485. The compliance must be assessed and certified by a Notified Body. Moreover, the Quality Management System must include Clinical Evaluation, Post Market Surveillance and Post Market Clinical Follow-up plans.
2: PREPARE THE TECHNICAL FILE
In the Technical File you have to include the information about the Intended Use of your Medical Device Software, all the testing reports as well as the Clinical Evaluation Report, your risk management plan and other information specified in Annex II of the MDR.
3: REGISTER IN THE EUDAMED DATABASE
Register your Authorised Representative who will handle the regulatory matters as well as your company as a medical device manufacturer in the EUDAMED database. It’s important to note that the authorised representative is mandatory only for importers outside of the EU.
You don’t necessarily have to do it at this point. It can also be done earlier or later in the process.
4: PREPARE A DECLARATION OF CONFORMITY
The next step for you, the manufacturer, is to prepare a Declaration of Conformity. It’s a legally binding document in which you affirm that your Medical Device Software is compliant with the MDR. At this stage, your product becomes officially CE marked.
5: REGISTER THE MEDICAL DEVICE SOFTWARE IN THE EUDAMED DATABASE
Now it’s time to register your software in the EUDAMED database. Keep in mind, that the UDI number must be placed, for example, in the “footer” or “About” section or other visible and easy-to-access places.
That’s it – your Medical Device Software is ready to be sold on the EU market. Your duty as a manufacturer is now to keep it safe and effective. In this regard, the EU MDR requires that manufacturers conduct Post Market Surveillance Activities. Also, each year you will undergo a Notified Body surveillance audit to ensure continuous compliance with the EU MDR. If you fail to pass it, your CE marking may become invalid.
During the transitional period, the manufacturers are required to perform the Clinical Evaluation and Post Market Surveillance Activities required by MDR, even though their CE marking certification for class I is still valid.
How to CE mark your software if it falls into a higher risk class?
The certification of software that belongs to a higher risk class requires the involvement of a Notified Body. A Notified Body shall audit the quality management system to determine whether it meets all the regulatory and product requirements. If the quality management system conforms to the relevant regulatory provisions, the notified body shall issue an EU quality management system certificate. After that, the declaration of conformity can be signed. Then, the product can be registered in the Eudamed database and finally introduced into the market.
Specific requirements depend on the characteristics and the level of complexity of a product as well as the risk associated with using it, which directly translates into the scope and timeframe of the audit.
How long is the CE marking certification valid?
The CE marking certification for classes IIa, IIb and III is valid for up to five years. After the five-year period, the registration of the certification must be renewed. It’s key to have it on the radar and plan the process in advance. It’s recommended to start the registration renewal at least six months before the certification expires.
How much does it cost to get CE marking certification?
The total cost of the CE marking certification process is proportionate to a device’s class. The higher the class, the bigger the risk and the more complex the process. Hence, the cost is also higher. The CE Marking certification is the most expensive for class III devices and can reach even tens of thousands of dollars.
How long does the CE marking certification process take?
Since there are still very few Notified Bodies, conducting the conformity assessment may take quite a long time. The time to begin a Notified Body audit varies between half a year and a year. In comparison, under the MDD it was usually a quarter. The time between an audit and approval may take up from two to even seven months.
Why so long? The EU MDR brought in more requirements, especially for the QMS. This makes the whole process more complex and time-consuming.
One of the reasons why it takes so long is also that there are few Notified Bodies that got already accredited in compliance with the new MDR requirements.
Currently, the MDR requires Notified Bodies to make their price list public. However, it’s worth noting that it includes the hourly rates. It’s especially important to keep in mind when introducing a product to the market for the first time (also for the first time since the MDR came into force) as it requires more than one audit to be performed. Firstly, the Notified Body checks the readiness for certification and after a few months it conducts the certification audit. It significantly impacts the cost. The yearly surveillance or recertification audits are performed once per year.
In total, the CE marking certification process may last more than a year. It’s important to take it into consideration when planning your business strategy. For example, you can optimise the process by creating an MVP first and start the CE marking Certification in the meantime, while at the same time developing your product further on.
Need support in the CE marking certification process?
We provide a technical file ready to be assessed for conformity by a Notified Body. Moreover, our specialists can conduct an independent conformity assessment before the official one to check what areas need improvement.
The software we create is compliant with all legal requirements, especially the IEC 62304 and ISO14971. In addition, we provide a Technical File and offer consultation and support with completing the CE marking certification process.
For more information, use the form at the bottom of this blog to contact Krzysztof Minicki, Director of Healthcare and Life Sciences.
As we enter 2023, Jama Software asked selected thought leaders – both internal Jama Software employees and our external partners – across various industries for the trends and events they foresee unfolding over the next year and beyond.
In the final blog of this five-part series, we asked Steve Neemeh, CEO / CTO of LHP Engineering Solutions – Danny Beerens, Senior Consultant at Jama Software – and Richard Watson, Practice Director at Jama Software – to weigh in on automotive product and systems development trends they’re anticipating in 2023.
Read more about the authors at the end of this blog.
2023 Predictions for Automotive Product Development
Design Trends – What are the biggest trends you’re seeing in your industry right now? How will they impact automotive product, systems, and software development?
Steve Neemeh: A generation ago software was introduced in engine controls that changed the automotive industry and allowed for efficiency and emissions improvement that mechanical systems could not provide. In today’s world, software is entering a new stratosphere of complexity. Rather than focusing on emissions, the goal is the user experience. High-tech meeting transportation changes the paradigm for automotive companies.
Danny Beerens: I don’t see a lot has changed in this regard. What is changing is what’s being built, not how it is being built.
Richard Watson: Taking advantage of Live Traceability™ will become increasingly important.
Definition of Live Traceability: The ability for any engineer at any time to see the most up-to-date and complete up and downstream information for any requirement, no matter what stage of development it is in or how many siloed tools and teams it spans. This enables the engineering process to be managed through data, and its performance improved in real-time.
Biggest Challenges – What are some of the biggest challenges you think automotive companies will be working to overcome in 2023?
Neemeh: The commercialization of the zero-emissions vehicle is the biggest challenge for 2023. The price points are a challenge. The supply chains are limited and not optimized for worldwide expansion. And, the energy grids are outdated in many places, such as California.
In terms of product and systems development, what do you think will remain the same over the next decade? What will change?
Beerens: More and more brands will move to electric vehicles, making those vehicles and specifically their motor management components more software driven. The various other components (primary functions, driver assistance/automation, as well as onboard entertainment) will also become more electronically controlled and thus software driven.
[Side note] Autonomous driving vehicles even sparked new fields in Software Engineering, like Ethical Software Engineering (studies the interactions of human values and technical decisions involving computing).
Clearly the Automotive Industry is shifting from Hardware/Mechanical Engineering and Electo-Mechanical to Software Engineering, forcing Product Data Management, or Product Lifecycle Management, vendors to start including Application Lifecycle Management into their environments. Hence you see Siemens Teamcenter has acquired Polarion and PTC Windchill acquired Codebeamer recently.
The Holy Grail will be an ALM/PLM environment that truly embraces Configuration Management for all engineering disciplines involved, combined.
Anticipating a new player not hindered by their already existing PLM or ALM application, neither their customer base, to develop a truly all incorporating ‘Engineering Assets Configuration Management’ environment, platform or application.
For the next decade, next to fully autonomous driving vehicles, flying cars might be the new way to fight congestion and a more personalized way to get around.
Regulations – What changing regulatory guidelines do you anticipate having an impact on companies in 2023?
Neemeh: With any new products in automotive, recalls will drive governments to regulate safety more closely. Functional safety is now a common term in automotive and most large OEMs are trying to find a way to comply and keep themselves from facing potential liability. The implementation of functional safety in the software development process will keep inching forward until a trigger makes it mandatory.
How do you foresee regulations shifting in Automotive Product and Systems Development over the next decade?
Beerens: Certainly, autonomous driving will introduce regulations to control not only functional safety and cybersecurity, but also for road safety (and legal responsibility) to interact with non-autonomous driven cars, until we’ve reached an era where none of us drive ourselves and all cars are controlled centrally to manage traffic flows.
Demands on alternative powertrains (e.g., hydrogen, or fuel cells) and existing electric driven cars’ necessity for fast charging and/or quick exchange of batteries, will spark off new technologies.
Apart from the obvious increase in data points and data exchange of the vehicle itself (sharing information for predictive maintenance, or usage of the car; tachograph in trucks) and its manufacturer and/or service station, G5 Connectivity of (autonomous driving) vehicles interacting with new traffic control instruments in, next to or on the road that assist with difficult traffic situations (automatically move to the side to allow emergency vehicles to pass), or location (purposely slow down at intersections that don’t have clear visibility of oncoming traffic) and react to traffic lights.
As a reaction to reduce CO2 emissions (cars sales are in a slow decline for a few years now already) new forms of mobility will arise where MaaS (Mobility as a Service) are being offered, sparking off disruptive newcomers to the traditional car sharing companies (renting: Hertz, and even taxi: Uber), like for example Lynk&Co, offering “memberships” for more flexible car usage and for car sharing with family and friends.
Tool Innovation – From an automotive engineering toolset perspective, what are some of the processes you think forward-thinking firms will be working to leverage or incorporate into their process and why?
Neemeh: Functional safety requires a strict development process and tools that support that process. Traditional tools only meet a small piece of that. They need to be integrated into an overall workflow and safety culture.
Any major disruptions to the Automotive Product and Systems Development industry you’re anticipating in 2023?
Watson: Political environment, supply chain issues, increased cost of specific items (such as chips). This increased cost is pushing the buyers into higher income areas, changing what kinds of cars will be successfully built.
Because of cost issues, refurbishing and retrofitting existing cars will become increasingly important. Similarly, car sharing will be increasingly wanted to control costs.
What role will cybersecurity play in automotive development in the coming year and beyond?
Neemeh: Safety can’t be achieved without cybersecurity. Assessment of your system’s vulnerability and its inclusion in your safety case is key to overall product acceptance. The more that cars become connected, the more this becomes important. Autonomous driving will be the pinnacle of connected cars. The more we move in that direction the more cybersecurity becomes a concern.
What sorts of process adjustments do you think development teams will need to make to be successful in 2023?
Watson: Automotive systems continue to have a stronger focus on software and this shift will continue. Different categories of software are provided in a vehicle from safety-critical to entertainment and this drives complexity sky-high.
With regulations continuing to get more stringent, development practices for non-safety-critical software systems must be tightened and this drives a focus to improve Agile practices. “Agile” is not an excuse to “throw something together” and must be supported by improved specification and verification techniques.
In your opinion, what are the biggest differences between an automotive company that survives to see 2030, and one that doesn’t?
Neemeh: Getting prototypes on the road and small-scale production with new technology (EV/Autonomous) is a monumental feat. The next step, however, is the commercialization of that technology into a transportation industry that is concerned about public safety. Those that consider that in the rollout and enable the scaling of safety-critical infrastructure will win, while the others will hit a brick wall of regulation.
Watson: A combination of sustainability with control of spiraling costs. There is a world shift in planetary awareness and the automotive market is at the forefront of reducing our consumption of fossil fuels. Car prices are increasing beyond inflationary rates and this increase will price out much of the lower market. Only organizations that can shuffle sustainability, quality and costs will survive this decade.
What role will cybersecurity play in automotive development in the coming year and beyond?
Watson: A shift towards Internet of Things (Iot) has exposed almost all aspects of automotive systems to the internet and social media. Cybersecurity will take a stronger focus, especially for those software systems that already interact with our social networking applications.
Beerens: Not only for our social networking applications; for long all systems utilizing the various onboard connections simply accepted instructions, without checking if that instruction was from a valid source. The infamous hack of a Landrover during Black Hack 2014 proved that. Encryption and intrusion detection are a good line of defense, but Zero-trust (or validating the source of the commands) Cybersecurity will be increasingly important for onboard systems from entertainment systems, connections like CAN, wifi, bluetooth or NFC, to motor management.
What advice would you give to new companies entering the automotive industry?
Neemeh: Get your workflows set up and your tools ready and optimized before you start throwing bodies at problems. Engineers are expensive. When they are set up properly, they can create miracles. But if they are burdened with administrative problems, they will get frustrated and leave.
Beerens: Look at established tool chains and industry templates to have a running start at the get-go. The European Union has an advisory board with such tool chains and templates. Concern yourself with compliancy from the beginning. Which compliancy standards you concern yourself with will depend on what parts of the auto you are working on.
Watson: Don’t try and define and invent the wheel and get help. There are many development tools available, find which tools work best based on tool reviews. Once selected, ask the vendor for the best way of working and don’t force the tool to do inefficient practices.
What topic(s) do you wish companies were paying more attention to?
Watson: Understanding how to address complex problems without the systematic nature we have relied upon. This is the only way to keep control of costs.
Predictions – What do you think will remain the same in your industry throughout 2023?
Neemeh: The adoption of electric vehicles will continue. Governments are behind it and the adoption rate is increasing.
What do you predict for regulation in the Automotive industry in 2023?
Neemeh: Involvement in the design process and review of ADAS features will become more important. The NHTSA has already started putting frameworks in place for that in the USA. In Europe, functional safety is commonplace and regulated already.
Will those trends still be prevalent 5 years from now? 10 years?
Neemeh: Yes, and it will move as fast as ADAS features move forward. Any autonomous Level 5 applications will jump-start this trend.
Where do you see Jama Software fitting in as the product development landscape evolves, and what can our customers expect as 2023 approaches?
Watson: Jama Software® is perfectly positioned to help the automotive industry allowing extended stakeholders to be directly involved with authoring and reviewing specification and verification activities rather than relying on tool super-users and PDF reports.
Beerens: Jama Connect® is a perfect fit for Product Design and collaboration with all its Stakeholders to refine, expand and improve Product Design, before any of these (proposed) changes are even visible in a PLM environment thereby preventing disruptions in Production before consensus has been reached.
——————————–
About the Authors:
Steve Neemeh joined LHP in 2015 to lead the expansion of the west coast operations. He is the leader of the strategy and solutions architects as well as president of the delivery consulting organization. Steve has over 25 years of Functional Safety experience prior to joining LHP. Steve has launched multiple start-up operations and has taken them to full production. Notably, a complete ground up electronics and software development group to service commercial aerospace electronics and military vehicle power electronics. For LHP, Steve pioneered the implementation of safety critical applications in California, launching functional safety for autonomous driving applications as well as air mobility.
Danny Beerens has 15 years of experience implementing, training, maintaining and supporting Application Lifecyle Management processes and their environments. Danny started in Software Configuration and Change & Defect Management (i.e., Workflows.) After joining Telelogic, he moved into Requirements and Test Management over the last decade, in roles as Support Engineer, Process Engineer, Consultant, and System Architect. Throughout his career Danny’s worked on projects and collaborated with customers in the Medical Devices, Aerospace & Defense, Automotive, and Semi-conductor industries. “The need to integrate ALM and PLM (and even beyond!) is apparent across all industries.”
Richard Watson is the Practice Director for horizontal solutions, engaged in identifying and creating services and assets spanning the Jama Software vertical solutions. Richard has a client first attitude and is passionate about Requirements and Systems Engineering. Based in the UK, Richard has been working in the systems and software industry for nearly 35 years and has been directly involved in most aspects of Systems Engineering from testing flight systems, through to software development of modeling tools, and Product management of IBM DOORS. Richard joined Jama Software as Practice Director in 2021.
Jama Software is always looking for news that would benefit and inform our industry partners. As such, we’ve curated a series of customer and industry spotlight articles that we found insightful. In this blog post, we share an article, sourced from Engadget, titled “TSMC is building a second chip plant to meet US semiconductor demand” – originally published on December 6, 2022, and authored by Steve Dent.
TSMC is Building a Second Chip Plant to Meet US Semiconductor Demand
The expansion marks one of the largest direct foreign investments in the US.
The White House and Taiwan Semiconductor Manufacturing co. (TSMC) have announced plans to build a second chip plant in Arizona, AZCentral has reported. That will boost the company’s investment in the state from $12 billion to $40 billion, while heavily reducing US reliance on semiconductor imports.
Both TSMC factories combined will produce 600,000 wafers per year. “At scale, these two [plants] could meet the entire U.S. demand for U.S. chips when they’re completed,” the National Economic Council’s Ronnie Chatterji told CNBC. “That’s the definition of supply chain resilience. We won’t have to rely on anyone else to make the chips we need.”
The newly announced factory will produce cutting-edge 3-nanometer chips by 2026. The expansion marks one of the largest direct foreign investments in the US and the largest in Arizona. TSMC recently upgraded its plans at its first facility as well, announcing it will now manufacture 4-nanometer instead of 5-nanometer wafers. The first chips are set to be manufactured there starting in 2024, with Apple and NVIDIA reportedly among the first customers.
The CHIPS and Science Act allotted $52.7 billion in loans and other incentives, plus billions more in tax credits, to encourage US semiconductor manufacturing investment. The legislation aims to boost private financing in chip manufacturing in the US.
President Joe Biden is set to visit the site of TSMC’s first plant later today, but the White House announced other related news yesterday. The US Department of Commerce and the European Commission are striking a deal to implement an “early warning mechanism” related to semiconductor chain disruptions following a pilot program last summer. The aim is to improve forecasting of semiconductor supply and demand to achieve a balance between the two.
At the same time, the EU and US are implementing a “transparency” mechanism around public support provided to the chip sector. In other words, one side won’t blindside the other with unexpected semiconductor subsidies that could put either at a competitive disadvantage. A similar issue came up during a recent visit by French President Macron, as EU leaders complained that the US Inflation Reduction Act was unfair to non-American companies.
As we enter 2023, Jama Software asked selected thought leaders – both internal Jama Software employees and our external partners – across various industries for the trends and events they foresee unfolding over the next year and beyond.
In the fourth part of our five-part series, we asked Shawnnah Monterrey, CEO at BeanStock Ventures – Romer De Los Santos, Senior Consultant at Jama Software – Vincent Balgos, Director of Medical Device Solutions at Jama Software – Michelle Wu, Medical Device Consultant at Wu Consulting – and Ivan Ma, Medical Device Program Leadership – to weigh in on medical device product development trends they’re anticipating in 2023.
Read more about the authors and their organizations at the end of this blog.
2023 Predictions for Medical Device Product Development
What are the biggest trends you’re seeing in the medical device and life sciences industry?
Shawnnah Monterrey: Biggest trends we are seeing include a rapid migration to the cloud this includes: IoMT, Digital Health, Digital Therapeutics and Big Data such as Genomics, Biotech, and Pharma.
We are seeing a rapid shift towards newly derived clinical insights using pre-existing data from existing medical devices, such as:
Companion diagnostics which combine a diagnosis outcome with a therapeutic and monitoring of that treatment
Digital therapeutics which use software ONLY to treat patients as opposed to a drug or instrument
Novel clinical insights where two or more measurements are combined to produce a clinical determination
AI based diagnostics which often consume numerous inputs that could be measured, demographical or even genetic to derive new clinical insights
Romer De Los Santos: Digital health continues to be a major source of growth as personalized medicine, wearable devices, and mobile health gain wider acceptance. Cloud computing, AI, and machine learning are improving patient outcomes by encouraging innovation and making personalized medicine possible. As these constantly evolving technologies continue to grow in complexity the regulatory framework around medical devices that incorporate them are also evolving to keep up.
For many years, medical device manufacturers secured their devices by disabling or designing out interconnectivity. The rise of electronic medical record keeping has forced manufacturers to support limited interconnectivity. They usually depended on security measures taken by their customer’s IT department as the primary risk control measure. That’s no longer acceptable in our interconnected world. The FDA requires manufacturers to consider cyber security threats and to design controls to reduce these risks as much as possible. This has led to developers having to learn more about threat modeling to limit touch points into their software and to creating plans on how to handle data breaches.
The 21st Century Cures Act amended the definition of a medical device to exclude certain software functions. The FDA intends to focus oversight on software functions that affect patient data and therefore pose the greatest threat to patient outcomes. Wise developers architect their software systems based on clearly defined software functions that can be individually evaluated for risk, leading to a reduction in the regulatory burden. Designing and documenting modular software facilitates re-use and therefore faster time to market for novel medical devices.
Michelle Wu: AI and Machine Learning: I continue to see AI and Machine Learning as a trend for 2023. Any pitch competition I attend includes multiple products that are incorporating AI or machine learning. There’s attention now on companies to look for and counteract bias in the data sets and algorithms.
Health equity: A spotlight on health inequities shines brighter since the pandemic and fortunately many companies are looking to do good and do well. Telehealth, remote patient monitoring, digital health apps, are the top areas of innovations that I see to address these disparities.
Vincent Balgos: The pandemic continues to drive the industry, regulators, and the market for COVID-19 related products and services, so I would expect continual development in these areas as new SARS-CoV-2 variants emerge, or other as other diseases arise.
Continual integration of medical life products, and interoperability amongst devices. As software to grows as a critical part of medical device industry, whether standalone SW or integrated with other components, there are many areas for 2023 innovation such as:
Software as a Medical Device (SaMD), Software in a Medical Device (SiMD)
Cybersecurity
Complex data analysis such as bioinformatics, genomic sequencing, imaging processing
Artificial Intelligence (AI) and Machine Learning (ML)
New or modified regulations (EU IVDR, EU MDR, and potential US VALID Act) continue to change the landscape in how medical device and life science organizations develop, manufacture, and maintain products.
The new FDA Computer Software Assurance (CSA) guidance that revisits validation in context of the current Computer System Validation (CSV) approach. Many medical companies are looking at this new risk-based approach to streamline their activities, documentation and outputs as the current standard practice can be complex and cumbersome.
Biggest Challenges – What are some of the biggest challenges you think medical device and life sciences companies will be working to overcome in 2023?
Monterrey: Two of the biggest challenges I see are: monetization and regulatory clearance.
Medical devices revenue models rely heavily on reimbursement from CMS which require a CPT code. Obtaining a new CPT code requires a significant investment and burden on the medical device manufacturer to provide clinical evidence which not only shows efficacy but also provides A reduced cost of care when compared to existing methods and treatments. We are seeing that digital therapeutics are struggling in this area. One strategy has been for digital therapeutics to partner with an existing reimbursed pharmaceutical via revenue sharing. But on the upside CMS has recently provided a new code which allows prescription digital behavioral therapy to be reimbursed as a medical benefit which is trailblazing the path for other digital therapeutics to follow.
While digital health applications that are intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease are medical devices and have been regulated by the FDA for many years, there has been new entrants in the recent years that have gone under the radar. With the recently issued guidance from the FDA on Clinical Decision Support Software, FDA attempts to make it clear which products are regulated medical devices, and which are not. This will slow the reduction in the barrier to entry as many digital health applications begin to play catchup.
Ivan Ma: The supply chain for components and materials continues to remain constrained. With lead times stretching well past 6 months, and sometimes getting close to 12 months. Programs should plan for contingencies and with expectations that milestones that require physical materials will be impacted by the last part in. Be wary of strategies that start early but require more total effort to execute.
In terms of product and systems development, what do you think will remain the same over the next decade? What will change?
De Los Santos: The need to ensure traceability between requirements, testing, risk, and design will continue to be important in the next decade. Changes in what is considered medical device software will lead to revised regulatory strategies by companies agile enough to take advantage of these changes. Documentation must become more modular to match the software they describe.
Balgos: Based on my past 17 years in medical product development, the time pressures to launch safe and effective products quickly to the market has always been a constant theme.
Many attempt the “faster, better, cheaper” approach, but schedule has always been the driver when comes to the project’s managements iron triangle (scope, budget, schedule). While this “faster, better, cheaper” approach may work for other industries, the medical field is especially constrained in that a patient’s safety is non-negotiable.
What will change is how companies adapt to the complexities of the regulation landscape, innovative technologies, and ever growing knowledge of diseases, illnesses, etc. The adaption for advanced tools, processes, and digitization of information will continue to grow industry as scientists/engineers evolve their practices.
What changing regulatory guidelines do you anticipate having an impact on companies in 2023?
Monterrey: In addition to FDA’s guidance on Clinical Decision Support Software there are a few other draft guidance in the works such as Computer System Validation (CSA), Cybersecurity, and AI.
Tools that are used to implement part of all the quality system require validation to ensure that the tool is fit for purpose and mitigates the risk of failures that could pose undetected harm in the medical product. We have seen many of our clients spend significantly more effort on validating tools that do not pose significant risk to their medical device than the medical device itself. FDA’s Computer Software Assurance for Production and Quality System Software draft (CSA) guidance provides great insight on how to take a risk-based approach when validating your tools.
Cybersecurity affects all products in development and on-market, regardless of if they are fully embedded or even connected. For medical devices manufactures that have many legacy devices on-market, this new guidance can pose a significant risk and cost.
Artificial Intelligence and Machine Learning (AI/ML) Software as a Medical Device Action Plan provides some additional insights into FDA’s current thinking behind AI. Although there is no current guidance from the FDA, AI devices continue to be cleared under existing guidance increasingly year or over.
Balgos: The US VALID ACT could have major disruption to lab developed tests (LDT’s) and how they are regulated in the US market. The additional restrictions may impact the growth of new tests, but provide additional oversight to help improve safety. This controversial topic has been a continual discussion point in industry, and that the new VALID ACT provides some additional clarification and guidance.
How do you foresee regulations shifting in medical device and life sciences over the next decade?
De Los Santos: There is a growing understanding among regulatory bodies that cloud computing companies are developing technology that will significantly improve patient outcomes.
Tool Innovation – From a medical device and life sciences engineering toolset perspective, what are some of the processes you think forward-thinking firms will be working to leverage or incorporate into their process and why?
Monterrey: From an engineering toolset perspective – finding automated tools that support the regulation and the team’s ability to be agile for the full development cycle will have a significant impact. Typically, we see our clients taking 6 to 18 months back tracking design activities in order to satisfy the FDA when the product is almost completed. If development is done in a more automated and iterative way – time to market can be significantly reduced, more predictable and lead to higher quality products.
Wu: Tools that make regulatory compliance more efficient. The best tools make it easy for companies to enhance, instead of hampering, their product development and business strategy.
Human centered design. While not a new concept, it is not universally practiced and incorporated. Those that do this well have medical devices that resonate with users and have better product adoption.
Ma: Requirements matter more than ever. Avoid building the wrong thing by keeping track of requirements and risks management using a tool like Jama Connect. If you are paper tracing, you’re operating in the 20th century.
Any major disruptions to the medical device and life sciences industry you’re anticipating in 2023?
De Los Santos: AI, machine learning, and cloud computing were instrumental in the response to the pandemic but have far bigger implications for improving patient health. As companies shift focus away from the pandemic, I expect more innovation around personalized medicine and clinical decision support software, both of which take advantage of these emerging technologies.
Balgos: The US VALID ACT could have major disruption to lab developed tests (LDT’s) and how they are regulated in the US market and industry.
What sorts of process adjustments do you think development teams will need to make to be successful in 2023?
De Los Santos: Development teams should take advantage of the guidance on software functions to improve the architecture of their code and their documentation. The sooner development teams create re-useable code and documentation building blocks, the better.
Balgos: Aligning with new regulations, such as the potential VALID ACT, and new FDA draft guidances such as CSA, Human Factors, and others
For the EU market, organizations need to start early. Notified Bodies engagement as the backlog continues to be longer than expected for re-certification for Medical Devices and IVD’s to the new regulations.
In your opinion, what are the biggest differences between a medical device or life sciences company that survives to see 2030, and one that doesn’t?
Monterrey: Companies that strive to maintain agility while being regulated leveraging tool automation as opposed to paper-based and stage gate processes will have a competitive advantage and higher chance of survival by having the ability to:
Address cybersecurity demands in an ever-changing eco-system
Derive new clinical insights using real-world data
Innovate by releasing product and features in more frequent cadences
Stay ahead of obsolesce issues
De Los Santos: The ability to organize software, hardware, and documentation into re-useable building blocks are key to winning in this kind of environment. You must be fast while maintaining a level of quality that ensures patient safety.
Ma: Products that bring true clinical value will win in the long run. The challenge is finding organizations and sources of capital that are methodical enough to identify true clinical value and have the grit and determination to stick with programs that take more than 5 years to reach human use.
Balgos: Adapting to the environment will be key for a company’s survival. Whether new regulations, innovative technologies, or another global event changes in how industry operates, companies that has the ability, resources, and willingness to pivot will likely survive.
What role will cybersecurity play in medical device development in the coming year and beyond?
De Los Santos: Cybersecurity is here to stay! The FDA requires device manufacturers to document how they handle cyber security threats and breaches. Companies can’t depend solely on risk control measures made by the customer’s IT department.
What advice would you give to new companies entering the medical device and life sciences industry?
Monterrey: Invest in tools, training, and infrastructure upfront and hire industry and technological experts to help you navigate the complexity of the cloud environment and regulated space.
De Los Santos: Take some time to define a simple design and development process. Don’t overdo it! You don’t get extra credit for adding extra process work. Use a risk-based approach to determine how much is too much.
Wu: Understand that the path to commercialization is much longer for a regulated medical device or therapeutic as compared to a consumer good.
Gain an appreciation for the regulations, what claims you want to make for your product, and how those two impact your timeline.
Human-centered design, including addressing diversity and inclusion, will differentiate your product from others.
Ma: A mentor told me that medical devices are a hard but worthwhile sport. Play the sport with the intent to bring positive clinical value to people everywhere. The rest, as they say, will take care of itself.
Balgos: Understand the market, regulations, and intended use of products/services and the associated risks.
Encourage good documentation practices early and consistently, as documentation is the lifeblood of the industry. Because if it wasn’t documented, it never happened.
What topic(s) do you wish companies were paying more attention to?
Monterrey:
FDA requirements pre-development – implementing a QMS and following a design process.
Customer needs – developing with the end user in mind.
Software as a profit center – focused on the revenue opportunity software can bring.
Tool validation – focus on value-add activities, if you’re spending more time and money validating tools that verifying your medical device you should revisit your QMS for inefficiencies.
De Los Santos: I wish companies would take a little more time cleaning up their processes. Where are you wasting effort? Putting band-aids on your development process costs you more in the long run. What is a working medical product with a poor or non-existent design history file? It’s a brick. It’s a very expensive brick that will require months of remediation work. Design documentation created after the fact is always poor and you’ll also have trouble retaining great engineers if they must spend months remediating documents.
Wu: Women’s Health: While women make up 51% of the population, less than 1% of VC funding is going to FemTech. With an estimated market size of $1.186 Trillion by 2027, the medical device industry is slowly taking notice of the unmet need and market potential of innovation focused on women. Consumer product goods, digital health, and diagnostics are top three product addressing issues unique to women, including menstruation, maternal health, and menopause1. It’s an under tapped area that continues to be prime for disruption.
What do you think will remain the same in this industry throughout 2023?
Monterrey: I think we will continue to see slow economic recovery as a result of the side-effects of COVID as it relates to supply chain, pivots, and lower year end earnings. The businesses that end up striving will be those who are focused on long term strategy as opposed to short term reactions to the economy. Reinvestment and patience will be essential to staying ahead competitively.
What do you predict for regulation in the medical device and life sciences industry in 2023?
Monterrey: There will be a watchful eye on cybersecurity, additional thinking around AI and significantly longer wait times for approval.
Wu: While not significant changes in regulation, the change to MDR and IVDR in the EU continues its impact to the industry, especially as companies’ previous MDD certifications lapse, but have yet to obtain their MDR certifications. As of a July 2022 MedTech Europe Survey Report, >85% of existing medical devices that had MDD certification have received MDR. And unfortunately, it is the patients and public that live in the EU that will be affected when they no longer have access to the same medical devices and diagnostics that they had previously. With the 13–18-month time-to-certification with MDR-designated Notified Bodies, nearly double the time historically needed, this influences the worldwide go-to-market strategy of companies.
Will those trends still be prevalent 5 years from now? 10 years?
Monterrey: Digital health applications will begin to dominate the market over traditional hardware devices with new and innovative, diagnostics treatments and therapies leveraging cloud, AI and real-world data. FDA trends over the next 5 to 10 years will move towards harmonization to reduce complexity and improve ease of use. The reduce wait times the FDA will continue to extend devices in the break-through designation and rely on the use certification bodies or 3rd party FDA reviewers like BeanStock Ventures.
Where do you see Jama Software fitting in as the product development landscape evolves, and what can our customers expect as 2023 approaches?
De Los Santos: When properly configured and coupled with a simple design control process, Jama Connect significantly reduces the documentation burden for our customers. In the same way that a good source code management system facilitates code reuse, Jama Connect facilitates re-use of requirements, test cases, and risk documentation. There have been some recent improvements to the Jama Connect that I’ve been requesting since I was a Jama Software customer. I hope people take time to take advantage of them.
Shawnnah Monterrey – CEO, Beanstock Ventures
20+ years’ experience in the medical industry, Shawnnah Monterrey knows a thing or two about guiding innovative products to market.
Prior to founding BeanStock Ventures, she obtained a bachelor’s degree in computer science from the University of California, San Diego and an executive MBA from San Diego State University, then went on to hold product development management positions across numerous global firms, including Illumina, Invetech, Medtronic and Carl Zeiss Meditec. Through this work, she continued to develop a passion for innovation in medical devices, life sciences, and biotechnology.
BeanStock Ventures
BeanStock Ventures is 1 of 9 FDA-accredited Third Party Review Organizations globally which provides software development and regulatory compliance products and services to minimize complexity, and reduce cost and time to market of innovative medical devices.
BeanStock Ventures has over 140 years of combined experience in software development for the healthcare and life science space.
833.688.BEAN (2326)
Michelle Wu – Principal Consultant at Michelle Wu Consulting
Michelle Wu is a senior leader with 20 years of experience in the medical device and life sciences industries with roles in executive leadership, product and process development, manufacturing, and quality. Michelle has a history of successful medical device product development, strategic planning and execution, building teams, process evolution, and managing organizational change. She values a collaborative and diverse, equitable, and inclusive environment, believing that diverse perspectives lead to the best ideas, more cohesive teams, and better results.
Ivan Ma
Ivan Ma has nearly two decades of experience in the medical device industry holding leadership and design positions spanning a wide range of medical devices; from single use devices and active implantables to complex surgical robotic systems. Ivan specializes in bringing early phase projects through development in preparation for FDA submission and human use by introducing balanced discipline to an inherently chaotic process.
Vincent Balgos
Vincent Balgos currently leads the Medical Solution at Jama Software. Prior to joining Jama Software, he worked in the medical device / IVD industry for over 17 years with roles in systems engineering, product development and project management. Vincent has successful history in launching new products to the global regulated market, and is experienced in product development, risk management, quality systems, and medical device regulations.
Romer De Los Santos
Romer De Los Santos has been developing software and firmware in the medical device industry since 1999. He is proud to have been involved in the development of a wide variety of medical devices including insulin infusion pumps, continuous glucose sensors, solid state mobile SPECT cameras, sequencers, liquid handling robots, and various IVD assays. He’s served in the roles of software developer, product owner, scrum master, internal auditor, systems engineer, software project lead, core team leader, and technical product manager before joining Jama Software as a senior consultant this past February.
In part 2 of our blog series, we cover the second half of our eBook, “A Guide to Road Vehicle Cybersecurity According to ISO 21434” – Click HERE for part 1.
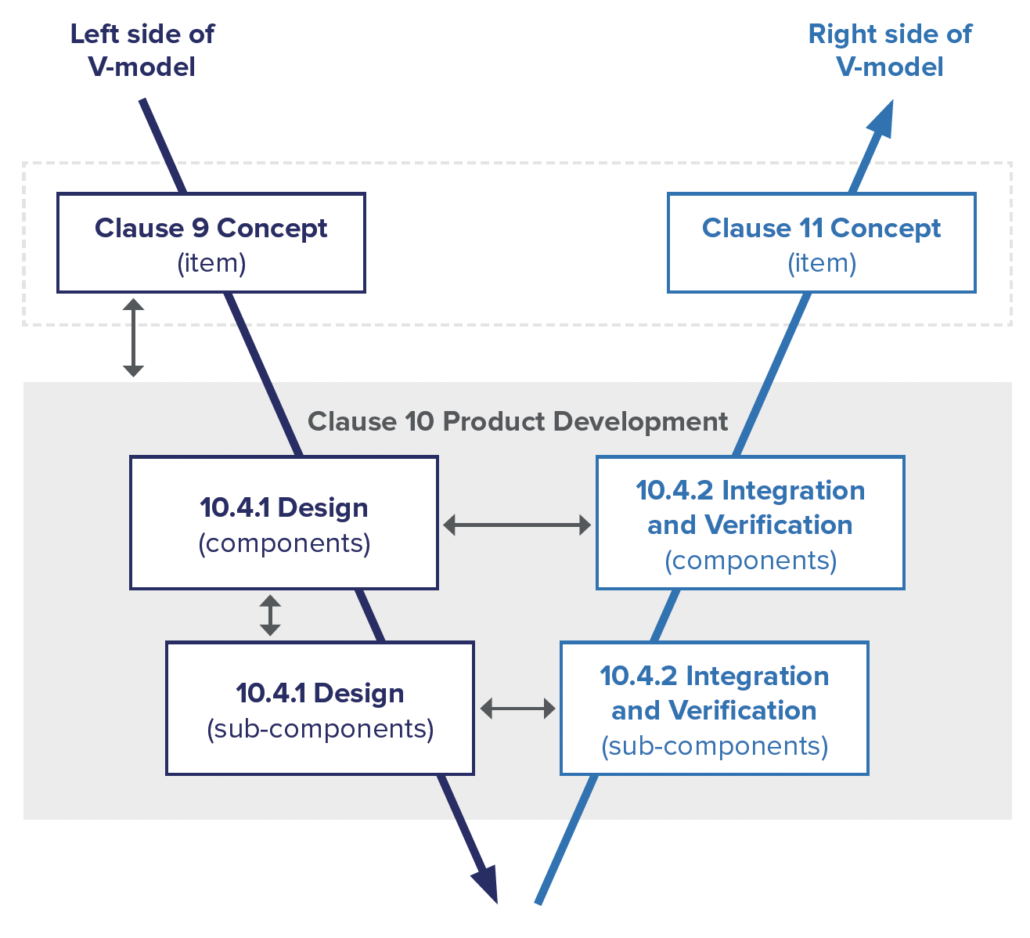
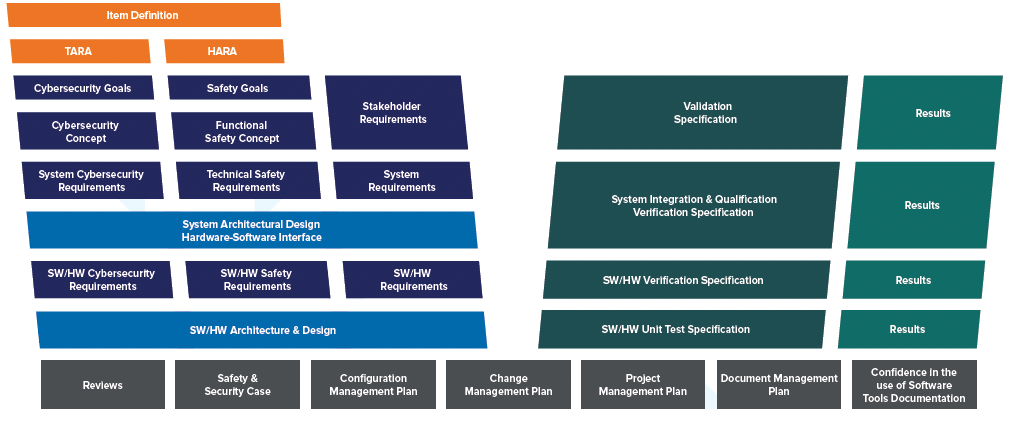
Much like other automotive standards, ISO 21434 defines a system engineering V-model to be followed for the development of cybersecurity features.
Concept Development
The cybersecurity V-model starts with the definition of the exact “item” that will be developed. The item is a component or set of components that implement functionality at the vehicle level and is defined in an item definition. In many cases, the same item definition may be used for both functional safety analysis and cybersecurity analysis.
Once the item has been clearly defined, a Threat Analysis and Risk Assessment (TARA) is performed to identify what cybersecurity threats exist for the item and what the risk of those threats are. For threats where the risk must be reduced, concept level requirements are developed, known as cybersecurity goals. Cybersecurity goals form the highest-level requirements for the system being developed from a cybersecurity perspective. For risks that will remain after cybersecurity goals are achieved, cybersecurity claims are documented to explain what, if any, risks still exist and why they can be accepted.
After defining cybersecurity goals, a cybersecurity concept is created. This documents the high-level concept that will be used to achieve the cybersecurity goals. The concept takes the form of cybersecurity requirements as well as requirements on the operating environment.
Product Development
Once a cybersecurity concept has been developed, the system must be designed in a way that will satisfy the cybersecurity requirements. Any existing architecture must be updated to consider the cybersecurity requirements. Each component of the system should be designed to support the cybersecurity requirements.
Although ISO 21434 provides an example of developing a system in two layers of abstraction, no specific number of layers is required. Instead, the standard leaves it to the product development organization to define a process appropriate for the complexity of their system. This ensures that organizations can adapt the standard to a wide range of systems and, for many, means that their existing system engineering process will satisfy ISO 21434.
Once the components of the system have been designed and integrated, the system must be verified to ensure that it meets the cybersecurity requirements.
The methods for verifying the system can include:
Requirements-based testing
Interface testing
Resource usage evaluation
Verification of the control flow and data flow
Dynamic analysis
Static analysis
The integration and verification activities should be documented in a verification specification and the results of verification documented in a verification report.
Validation of Automotive Cybersecurity Goals
While the focus of verification is ensuring that the item meets the cybersecurity requirements, validation ensures that the item achieves the cybersecurity goals. This is done by first validating that the cybersecurity goals are adequate and then validating that the item achieves the cybersecurity goals. Validation may involve reviewing work products, performing penetration testing and reviewing all the managed risks previously identified. A rationale for the validation activities is required. The completed validation is documented in a validation report.
Even after product development is complete, the cybersecurity lifecycle continues.
Production
During the production phase, the item that has been developed is manufactured and assembled. A production control plan is required to ensure that cybersecurity requirements for post-development that were identified earlier in the lifecycle are applied to ensure that no vulnerabilities are introduced during production.
Operations and maintenance
Once an item has been integrated into a vehicle and the vehicle is on the road, new cybersecurity threats can still be identified. ISO 21434 requires organizations to have a plan for how to respond to this scenario.
Organizations must create a cybersecurity incident response plan each time a new cybersecurity incident occurs. This plan includes what remedial actions are required and how they will be performed. The response may range from providing new information to vehicle owners, to over-the-air updates, to recalls where the owner must bring the vehicle in for service.
End of cybersecurity support and decommissioning
Given that the cybersecurity lifecycle continues after vehicles have been sold to consumers, a method for ending cybersecurity support for those vehicles is needed. ISO 21434 focuses on developing a plan for communicating with customers when cybersecurity support ends. Since decommissioning can occur without the organization’s knowledge and in such a way that decommissioning procedures cannot be enforced, ISO 21434 only requires making documentation available to explain how to decommission the item with regards to cybersecurity, if this is even required.
Integrating the Automotive Cybersecurity with Overall System Engineering
ISO 21434 defines many cybersecurity-specific requirements and requires personnel with specific cybersecurity knowledge and skills. Because of this, it may be tempting for organizations to silo cybersecurity engineering activities from other engineering activities, but this would be a mistake. While risk analysis required by ISO 21434 can be considered as a separate activity from other system engineering activities, a single product still must be developed that meets a wide range of requirements, including cybersecurity requirements. For this reason, it is best to manage a unified database for requirements, architecture, and design, rather than tracking cybersecurity artifacts separate from others.
To support this, think of cybersecurity analysis as another input to product development, just like functional safety analysis and market analysis.
By taking a unified approach, a single system engineering V-model can be implemented that describes an overall product development process that incorporates cybersecurity without creating silos. While specialists will be focused on performing cybersecurity analysis, implementing known best practices and validating the final system achieves cybersecurity, this must be done in cooperation and coordination with the rest of product development.
How Jama Connect® Supports Cybersecurity Engineering
One way to implement a unified requirements, architecture, and design database is by using Jama Connect®. Jama Connect for Automotive provides a framework that incorporates the key requirements of ISO 21434 into a single project structure along with overall system engineering.
Specifically, Jama Connect for Automotive provides guidance on supporting the following activities:
TARA Cybersecurity goals
Cybersecurity concept
Design Integration and verification
Validation
An example of the framework is shown below:
Conclusion
ISO 21434 introduces a robust framework for organizations to apply the state-of-the-art in automotive cybersecurity to their product development. This framework is necessary from both a market and regulatory perspective. The high-level of connectivity available in vehicles today means that there many ways for someone to maliciously change a vehicle’s operation. While many consumers may be unaware of the risks today, if there are ever accidents that result from cyber-attacks, that will change quickly. A vehicle OEM’s brand will surely be impacted by such as incident. In addition, regulators have already imposed strong cybersecurity requirements in many regions. ISO 21434 is quickly becoming an essential regulation for companies developing products at all levels of the automotive supply chain.
Whether your team is young or seasoned, small, or large, all together or scattered across boundaries, Jama Connect for Automotive can help improve processes, reduce costs, improve time to market, and help achieve ASPICE compliance. To learn more about Jama Connect for Automotive, download our datasheet.
Interested in learning more about how Jama Connect for Automotive can help provide your team meet market demands more quickly and efficiently?
According to this EU survey, 17,095 valid medical device and IVD certificates are set to expire in 2024 and 2025. Now is the time to get your requirements management and documentation in order, so you’re in the best position to meet with notified bodies and update your certification.
Euro Roundup: MDCG publishes guidance on MDR, IVDR authorized representative requirements
The Medical Device Coordination Group (MDCG) has published guidance on the role and requirements of authorized representatives under the new medtech regulations. In the guidance, MDCG unpacks what the Medical Devices Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) mean for authorized representatives, manufacturers and other economic operators.
EU regulations require manufacturers without a presence in a member state to appoint a sole authorized representative who serves as their EU contact person and is central to ensuring compliance . MDR and IVDR outline the obligations of authorized representatives and enhance their responsibilities.
“The manufacturer and the authorized representative are free to configure the structure of their contractual relationship as they see appropriate, as long as there is a written mandate that meets the minimum requirements of Article 11(3) of the Regulations and the content of which is agreed between the parties. A mandate should be drawn up irrespective of whether the authorized representative is independent/outside of or is part of the same larger organization as the manufacturer,” according to the guidance.
Article 11(3) requires the authorized representative to perform tasks specified in their mandate with the manufacturer. Upon request, the authorized representative must provide a copy of the mandate to the competent authority. The article also describes the minimum tasks that the mandate should cover, but the authorized representative can agree to take on additional activities. Certain responsibilities cannot be delegated by manufacturers to authorized representatives.
The responsibilities of authorized representatives include verifying the existence of EU declaration of conformity and technical documents and, if applicable, that an appropriate conformity assessment procedure has been carried out by the manufacturer. Authorized representatives may inform their manufacturers if they have reason to believe the conformity assessment procedure is inappropriate for the device in question. Other tasks include keeping a copy of the EU declaration of conformity.
Authorized representatives need to comply with the registration obligations set out in MDR and IVDR, for example by registering their details in EUDAMED. The regulations require authorized representatives to update their information within one week of a change.
Three pharma trade groups have warned that EU proposals on wastewater treatment will jeopardize access to medicines without helping the green transition.
The Commission sees applying a “polluter pays” policy to the pharmaceutical and cosmetics industries as a fair response to evidence that the sectors “are jointly responsible for 92% of the toxic load in wastewaters,” notably because there is “sufficient evidence on the existence of micropollutants from these products in wastewater and there are treatments to remove their harmful residues.”
AESGP, EFPIA and Medicines for Europe, trade groups that represent different types of drugmakers, see things differently. In a joint statement, the bodies branded “blanket levies on medicinal products based on patient excretion levels” as “unprecedented, disproportionate, unfair and ineffective.” The trade groups warned the proposal will be “very detrimental to society, if increased burdens on companies mean that many essential medicines are no longer viable and result in shortages.”
“It is frustrating that duplicative and unworkable proposals for levies on medicines to address wastewater management are thrown at the pharmaceutical industry. This undermines our efforts to reduce our environmental footprint and more worryingly, negatively impact patients who need medicines. It is a lose-lose proposal,” said Adrian van den Hoven, director general at Medicines for Europe.
Few active pharmaceutical ingredients pose risks to the environment and “these are very well under control given the concentration levels found in European waterways,” according to the joint statement. As “pharmaceuticals are only a small fraction of the substances that an improved wastewater treatment would remove,” the proposal would see drugmakers finance wastewater treatment upgrades that would remove micro-pollutants from other “unaccounted” sources of water contamination.
PRAC recommends restricting use of JAK inhibitors in inflammatory disorders
The Pharmacovigilance Risk Assessment Committee (PRAC) has proposed restricting the patients who receive JAK inhibitors for chronic inflammatory conditions to mitigate risks linked to the molecules.
JAK inhibitors including AbbVie’s Rinvoq and Pfizer’s Xeljanz are used to treat inflammatory conditions such as rheumatoid arthritis. However, regulators are wary of the side effects caused by the class of molecules, leading the US Food and Drug Administration to apply boxed warnings and PRAC to review the EU rules to ensure the benefits outweigh the risks.
The European Medicines Agency (EMA) committee recommends that patients aged 65 years or above, people at increased risk of major cardiovascular problems, individuals who smoke or have done so for a long time in the past, and those at increased risk of cancer should generally receive other treatments. Those patients should use JAK inhibitors only in the absence of suitable alternatives.
PRAC’s proposed restrictions reflect the findings of a clinical trial of Xeljanz, which linked it to a higher risk of major cardiovascular problems, cancer, venous thromboembolism, serious infections and death than TNF-alpha inhibitors. Some developers of JAK inhibitors have sought to differentiate their molecules based on safety, but the PRAC recommendation covers five drugs for chronic inflammatory disorders.
Notified body survey quantifies the 2024-25 bottleneck for medical device, IVD certifications
An EU survey has revealed the number of medical device and IVD certifications that are set to expire in 2024 and 2025, respectively. Notified bodies have identified the years as a potential bottleneck in the transition to MDR and IVDR.
While the EU has countered the threat of MDR and IVDR causing near-term supply disruption, the delays have potentially only pushed the problems out by a few years. The EU survey of notified bodies shows that 17,095 valid certificates issued under the old device directives will expire in 2024, compared to 1,387 this year and 4,311 in 2023.
As of October, companies had filed 8,120 MDR applications, up from 6,188 in April. Applications are growing faster than MDR certifications, which rose from 1,069 in April to 1,990 in the latest survey.
EDQM strikes agreement with EU to support substances of human origin regulatory framework
The European Directorate for the Quality of Medicines & HealthCare (EDQM) has agreed to enhance its cooperation with the EU on substances of human origin (SoHO) such as blood, organs, tissues and cells.
Under the terms of a jointly financed agreement that will run to 2024, EDQM will “contribute to providing all Council of Europe member states, including the EU 27, with a coherent European regulatory SoHO framework and to supporting professionals of the sector in implementing this framework and in strengthening their SoHO systems.”
The agreement builds on decades of collaboration with the EU, including technical cooperation on SoHO that dates back more than 10 years. EDQM and the EU framed the expanded scope of the agreement to make “the best use of their respective strengths and resources.”