
One of the challenges I often hear from our automotive customers is that a large portion of time disappears into tasks that are both very manual as well as very tedious.
An example of one of these tasks is scanning standards or regulatory documents, finding consistencies both between the standards and their internal requirements, then tracing them down to relevant system requirements or generating newly needed requirements from them.
There is much that can be gained by bringing AI agent toolkits into the equation, while keeping key judgement points in human hands.
Two technologies come together to optimize this workflow:
- Independently built agent toolkits that are specifically for automotive engineering workflows.
- Model Context Protocol (MCP) built specifically to give governed access to requirements and traceability within Jama Connect®.
Putting these together gives teams the capability of taking AI generated content from standards all the way to auditable and traceable artifacts, without the need of a manual data entry gap or consistency check in between.
Dedicated AI Agents Know the Standards
The automotive safety and security domain is very well suited to AI assistance and augmentation as so many of the workflows are very structured and driven by standards and regulation. Take standards like ISO 26262, ISO 21434, ASPICE, ISO 21448 (SOTIF) and you have a strong definition of specific work products with defined content requirements.
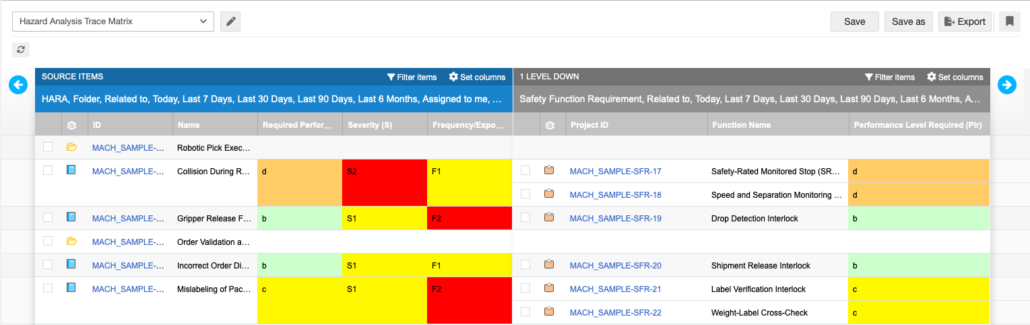
A Hazard Analysis and Risk Assessment (HARA) or Threat Analysis and Risk Assessment (TARA) has defined structures. Safety goals are asked to have specific attributes. Functional Safety Requirements (FSRs) derive from safety goals in predictable ways.
AI agent toolkits are being developed to take that structured knowledge and turn it into specialized agents who can do the analysis work for you.
Instead of spending time scouring the standards to build the needed requirements for a function, create a safety engineer agent for ISO 26262, a cybersecurity analysis agent who understands ISO 21434, an agent who understands the autonomous safety standards, and have them work together to build the necessary requirements and traceability for that function.
The AI takes care of the mechanical heavy lifting of standard analysis, formatting compliance, and first-draft generation. The SME engineer then applies contextual judgment, domain expertise, and accountability that no AI system can replace, particularly in a safety-critical domain where the engineer’s professional and legal responsibility is necessary.
The toolkit acts essentially like a knowledgeable engineering colleague who never forgets the standards but leaves the final verdict to the SMEs who are responsible.
This content could be created and then manually copied over into Jama Connect or fed through the API, but these introduce unnecessary bottlenecks when the process could be streamlined directly through an MCP.
Jama Connect Via MCP
Model Context Protocol (MCP) is a standardized interface that allows AI systems to interact directly with external tools and platforms. An MCP server for Jama Connect exposes the platform’s capabilities, such as creating items, updating attributes, establishing trace links, and querying project structure as operations that an AI agent can invoke directly during a workflow.
This adds context. Instead of analyzing a function in the abstract, the AI has direct access to the project’s actual requirements structure, existing items, coverage relationships, review statuses, and gaps in the current traceability graph, which changes the quality of the analysis.
Use Case: Hazard Analysis
An AI suggesting candidate hazards for a function is more useful when it can see which system-level requirements already exist in the project, which hazards have already been identified, and where coverage links are currently missing. The suggestions are grounded in the project’s living state rather than a generic interpretation of the standard.
The toolkit can now generate the content and trace it automatically in the predefined structure within Jama Connect, and then it’s up to the engineer to review the content and give approval throughout the process.
Here is what this might look like for a HARA workflow:
- The engineer invokes the safety agent with a description of the function under analysis. For example: an automated emergency braking system operating in urban environments at low speeds.
- The agent applies ISO 26262 Part 3 methodology to produce a structured hazard identification, severity/exposure/controllability ratings, ASIL determinations, and candidate safety goals.
- Via the Jama Connect MCP, the agent creates the HARA items directly in the live Jama Connect project, populated with the generated content, and associated with the appropriate upstream items.
- Safety goals are then created as downstream items, with coverage relationships to the HARA hazards already established.
- Any items created through this assisted workflow are labeled as AI-assisted drafts in Jama Connect so that the engineer can quickly filter and review them.
- The engineer reviews the generated content in Jama Connect, modifies what needs modification, and formally sends the items for approval, triggering the review workflow that the project’s functional safety process requires.
What AI should not be doing in a regulated safety context is determining final ASIL classifications, asserting compliance completeness, or producing artifacts that are treated as authoritative without explicit engineer review and approval. The professional and legal accountability for safety analysis outputs sits with the engineer, and the tooling should make that accountability concrete rather than diffuse it.
The source and history of every item visible and auditable, a requirement for any safety process that may face external assessment. An item created through AI-assisted analysis is not inherently less valid than one created entirely manually, but it must be traceable as such, and it must carry evidence of an engineering review before it can be treated as authoritative.
Move Faster With AI Agent Toolkits and Jama Connect MCP
What once took hours or even days can now be compressed into minutes, with effort focused on reviewing and refining results rather than manually creating them from scratch.
Engineering time is concentrated where it delivers the most value: applying expertise, judgment, and accountability instead of performing repetitive administrative tasks.
That speed comes with a clear accountability structure. AI agent toolkits do the analytical heavy lifting but every item that enters the authoritative record does so because an engineer reviewed and approved it. Items created via MCP are labeled as AI-assisted drafts in Jama Connect and cannot be treated as authoritative until a formal review is completed. That boundary is what makes this approach defensible in regulated environments and auditable.
For teams already using Jama Connect, the MCP approach is also complementary to the AI capabilities being built directly into the platform through Jama Connect Advisor™. Advisor supports in-application AI assistance such as analyzing requirement quality against INCOSE and EARS standards, generating test cases from requirements, as well as surfacing missing traceability links through relationship discovery. The MCP extends that reach further, allowing externally configured agents to operate across broader project context and bring standards-specific domain knowledge into the same governed workflow. The two layers work on the same system of record, so the governance model stays consistent regardless of where the AI assistance originates.
As AI agent toolkits become more capable and MCP adoption grows across the engineering toolchain, the scope of what can be analyzed, suggested, and routed for human review will expand significantly. Teams that establish the governance model now will be better positioned to absorb this capability without compromising the process integrity that safety-critical development demands. The goal is not to automate engineering judgment out of the loop. It is to give engineers more leverage over the parts of their work that genuinely require it.
Learn More About AI in Automotive Engineering
As AI becomes more capable, automotive organizations face a broader challenge: how to adopt AI at scale while maintaining governance, traceability, functional safety, and cybersecurity compliance.
To explore how leading automotive teams are navigating these challenges today, watch our on-demand webinar, How AI-Driven Engineering is Transforming Automotive Development, where I discuss practical strategies for balancing AI-driven innovation with the rigor required for safety-critical development.

? Definition and FDA Rules")






? How It Works and When to Use It")
