Understanding FDA Medical Device Class and Classifications, and its Impact on Requirements Management
 |
|
|
|

One of the early steps I advise my clients to take when developing their medical device is to determine the class and classifications of their medical device. In conjunction with the complexity of the device, understanding the class and classification sets the foundation for your product development timeline and effort.
This post gives a basic introduction to FDA medical device classes and classifications and the implications for your product development schedule and requirements management.
The FDA established three regulatory classes based on the level of control necessary to assure the safety and effectiveness of the device. Classification is based on the intended use of the device and indications for use, as well as the risk the device poses to patients and users.
There are three classes: Class I, Class II, and Class III. Class I devices are those with the lowest risk, Class II devices have a greater risk, and Class III includes devices with the greatest risk.
The FDA also established classifications for over 1,700 generic types of medical devices and grouped them into 16 panels, or medical specialties. Example panels include Cardiovascular Devices and Radiology Devices. Each of the generic types is assigned as Class I, Class II, or Class III.
The class and classification of the device impacts what FDA premarket submission or application is required for clearance to market. The common premarketing submission or application for each class are:
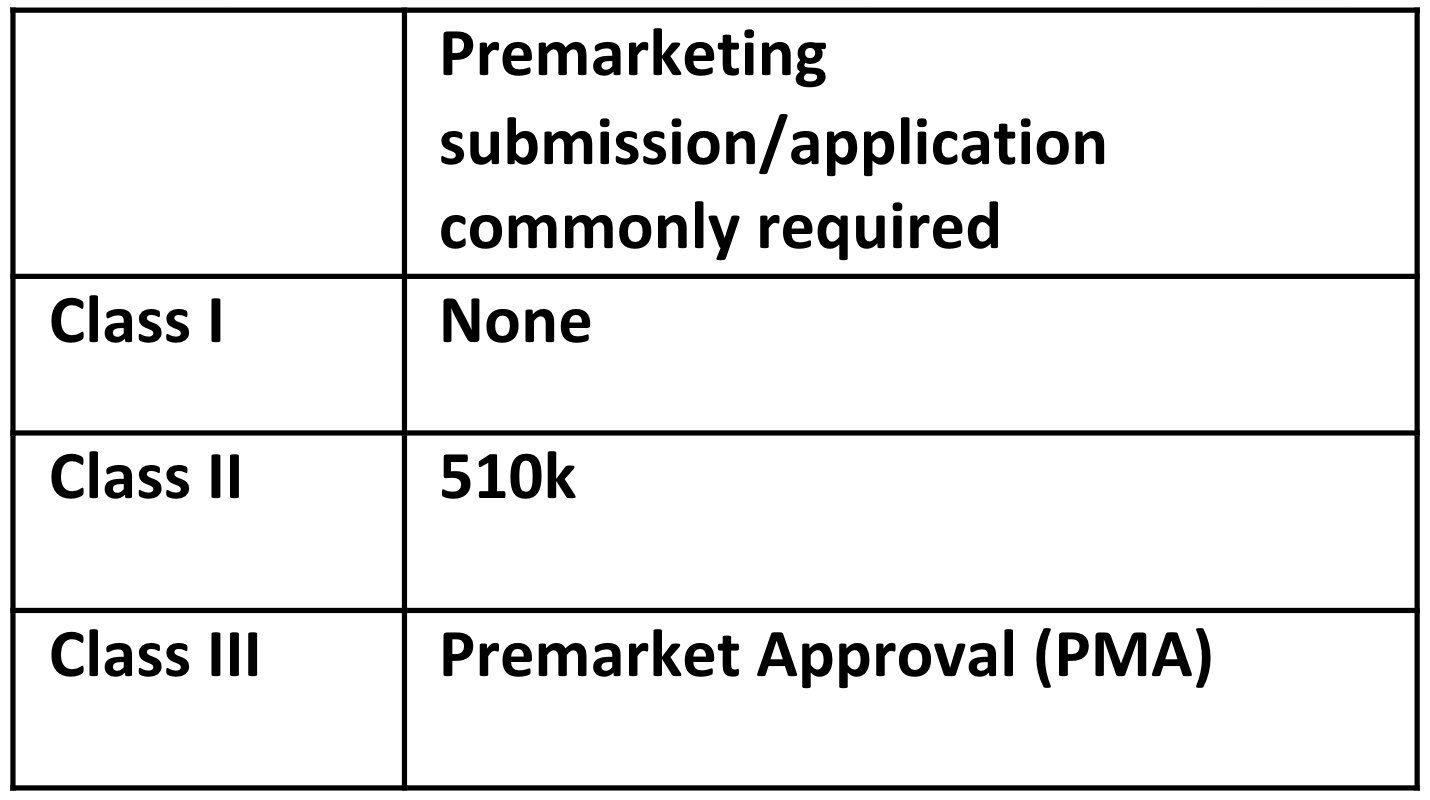
Note: These are the common regulatory submission and applications for each class of device. There are exemptions, limitations on those exemptions, special controls that may apply, and exceptions, so be aware whether any of these applies to your device. For example, about a quarter of Class I devices are not exempt, and a 510k premarket submission is required.
As the process for the 510k submission is 30-90 days, and the process for the more in-depth PMA submission is 180 days to accept or reject, this time should be understood and planned into your product development schedule.
Similarly, expect elements from the required design control process and design history file to be included as part of a 510k and PMA. Also keep in mind that when design controls are required for your device classification, the full design history file can be scrutinized as part of an FDA inspection of your organization. Since the FDA evaluates whether a device is effective and ensures the risk to the patients and users is appropriately addressed, good requirements and risk management is key. It’s important to have an organized manner in which to demonstrate and document that risk management and user needs are successfully traced through design inputs, design verification, and design validation. A requirements management tool like Jama Connect™ allows for this traceability in an efficient, collaborative, and regulatory-compliant manner.
Understanding your device class and classification is a key step to understanding the path for FDA regulatory clearance and subsequent design control requirements for your medical device development. Knowing those expectations up front will make for a smoother medical device development journey.