Notable Changes in the new FDA Draft Guidance – Content of Premarket Submissions for Device Software Functions
The FDA released its new draft guidance for the Content of Premarket Submissions for Device Software Functions on November 4, 2021. Once approved, this guidance will replace the preexisting Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices released in May 2005. A lot has changed in the last sixteen years where medical devices and software is concerned; the FDA released the new guidance citing technological advancements in all facets of health care, which has caused software to become an important part of many products and medical devices. The most impactful changes that we should be aware of from the guidance are as follows:
Change from Software Level of Concern (Major, Moderate, Minor) to Documentation Level of Concern (Basic, Enhanced)
Expanded scope, including –
Introduction of the term software function
Introduction of Software as a Medical Device (SaMD)
Addition of DeNovo Classification Request and Biologics License Application (BLA) to the premarket submissions to which the guidance applies
Introduction of additional consensus standards and guidance documents, including –
ANSI/AAMI/IEC 62304: Medical Device Software – Software Life Cycle Processes
Off-The-Shelf Software Use in Medical Devices
Provision of Examples (Documentation Level, System and Software Architecture Diagram Chart Examples)
Let’s analyze each of these changes and determine how impactful the change will be.
Where the change from Software Level of Concern to Documentation Level is concerned, there are several important things to consider when analyzing, the foremost being that Software Level of Concern is no longer used. The FDA now describes it as Documentation Level, which is more germane to the information conveyed. Additionally, the Documentation Level name change also avoids confusion with the IEC 62304 Software Classification and has the benefit of simplifying the process for determining the documentation level. Limiting the documentation level to two levels streamlines the process, as the only differences between the Moderate and Major Software Levels of Concern were the requirement to provide a Configuration Management Plan and Maintenance Plans instead of summaries, and the addition of Unit Test and Integration Protocols for the Major level of concern. Other changes related to the documentation to be provided are generally quite subtle, such as requiring the Risk Management File rather than just Device Hazard Analysis as specified by the current version of the guidance, and what used to be Software Development Environment Description being redesignated Software Development and Maintenance Practice. These changes create a tighter alignment with IEC 62304, highlight the importance of risk management and simplify the use of the guidance.
Although the scope of what the guidance is intended to cover remained unchanged, there are three new terms discussed in the scope section which serve to emphasize areas of importance to the FDA – device software function, Software in a Medical Device (SiMD) and Software as a Medical Device (SaMD). The term device software function is used to highlight the fact that a product could have multiple functions, a function being a distinct purpose of the product. SiMD and SaMD are new terms used in the industry, although the types of software covered by each term were already present in the original version of the standard. The premarket submissions for which the guidance applies now included the De Novo Classification Request (on July 9, 2012, FDA allowed a sponsor to submit a De Novo classification request to the FDA without first being required to submit a 510(k)) and Biologics License Application (as of March 23rd, 2020, all biological products must be approved through the BLA pathway), both of which were introduced after the current version of the guidance document. These changes serve to modernize the terminology used in the guidance and highlight the importance of the guidance being applicable to SaMD.
Another important update in the guidance is the mention of IEC 62304. This standard was first issued after the original FDA Guidance was released in May 2005; in the draft guidance, the FDA explains the relationship between its own draft guidance and IEC 62304. IEC 62304 is a software lifecycle process, whereas the scope of the FDA draft guidance is only the facilitation of appropriate review of premarket submissions, allowing for the evaluation of safety and effectiveness of medical devices. Since IEC 62304 is an FDA-recognized consensus standard, the FDA is allowing a Declaration of Conformity to IEC 62304 as documentation for Software Development and Maintenance Practices required by the draft guidance. In addition to the use of IEC 62304, the draft guidance also references other FDA guidance documents related to Cybersecurity and off-the-shelf software, which were likewise not available when the original version of the guidance was published in 2005. These changes also help modernize the guidance and demonstrate the importance of IEC 62304, as well as providing an indication of key areas that the FDA will expect to be addressed in future 510(k) submissions.
A further significant addition to the draft guidance is the inclusion of examples related to Documentation Level and System and Software Architecture. The examples included in the guidance provides different scenarios to show how to arrive at the Basic or Enhanced documentation levels. The draft guidance also provides an example of an architectural decomposition that can be applied to a system or software. In showing these diagrams, the FDA is proposing a level of documentation in the architecture document unseen in other FDA software-related guidance documents. The examples include different “functions” and are inclusive of SOUP (Software of Unknown Providence) modules. The inclusion of these examples provides an indication of FDA expectations when it comes to the documentation of software architecture and ensures that the new approach to determine the level of documentation required is well-understood.
In summary, the FDA Draft Guidance has been updated to bring it up to date with current standards and other guidelines that have been released since the initial guidance. There has been some level of simplification by substituting the software level of concern with basic and enhanced levels of documentation. Additionally, the guidance addresses current hot topics such as SaMD and IEC 62304. Examples have been added to the guidance to make it easier for users to understand and implement correctly.
RELATED
In this post, we highlight and summarize parts of the new “Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions Draft Guidance for Industry and Food and Drug Administration Staff.” It is important to note that this content contains non-binding guidance and therefore is not currently for implementation.
With that said, while this draft is non-binding, some organizations may adapt to this guidance proactively to address hot topics not covered in formal regulation, for example: U/X Human Factors, Software as a Medical Device (SaMD), combination products, and others.
This blog post, while quoting the FDA draft guidance directly, is not a full representation of the content. It is comprised of snippets that were particularly relevant or interesting to the author. It also does not cover any appendices.
Following this information, subject matter expert, Vincent Balgos, weighs in on the draft guidance. Read to the end to see his thoughts!
FDA Guidance for Cybersecurity for Medical Device
With the increasing integration of wireless, Internet- and network-connected capabilities, portable media (e.g., USB or CD), and the frequent electronic exchange of medical device related health information, the need for robust cybersecurity controls to ensure medical device safety and effectiveness has become more important.
In addition, cybersecurity threats to the healthcare sector have become more frequent and more severe, carrying increased potential for clinical impact. Cybersecurity incidents have rendered medical devices and hospital networks inoperable, disrupting the delivery of patient care across healthcare facilities in the U.S. and globally. Such cyberattacks and exploits may lead to patient harm as a result of clinical hazards, such as delay in diagnoses and/or treatment.
Increased connectivity and interoperability have resulted in individual devices operating as single elements of larger medical device systems. These systems can include health care facility networks, other devices, and software update servers, among other interconnected components. Consequently, without adequate cybersecurity considerations across all aspects of these systems, a cybersecurity threat can compromise the safety and/or effectiveness of a device by compromising the functionality of any asset in the system. As a result, ensuring device safety and effectiveness includes adequate device cybersecurity, as well as its security as part of the larger system. For the current edition of the FDA-recognized consensus standard(s) referenced in this document, see the FDA Recognized Consensus Standards Database.
Scope: Who Does This Apply To?
This guidance document is applicable to devices that contain software (including firmware) or programmable logic, as well as software as a medical device (SaMD). The guidance is not limited to devices that are network-enabled or contain other connected capabilities. This guidance describes recommendations regarding the cybersecurity information to be submitted for devices under the following premarket submission types:
Premarket Notification (510(k)) submissions;
De Novo requests;
Premarket Approval Applications (PMAs) and PMA supplements;
Product Development Protocols (PDPs);
Investigational Device Exemption (IDE) submissions; and
Humanitarian Device Exemption (HDE) submissions.
General Principles
This section provides general principles for device cybersecurity relevant to device manufacturers. These principles, found throughout this guidance document, are important to the improvement of device cybersecurity and, when followed, are expected to have a positive impact on patient safety.
These general principles include:
Cybersecurity is Part of Device Safety and the Quality System Regulations
The documentation recommended in this guidance is based on FDA’s experience evaluating the safety and effectiveness of devices with cybersecurity vulnerabilities. However, sponsors may use alternative approaches and provide different documentation so long as their approach and documentation satisfies premarket submission requirements in applicable statutory provisions and regulations. The increasingly interconnected nature of medical devices has demonstrated the importance of addressing cybersecurity risks associated with device connectivity in device design because of the effects on safety and effectiveness.
Cybersecurity risks that are introduced by threats directly to the medical device or to the larger medical device system can be reasonably controlled through using an SPDF.
The primary goal of using an SPDF is to manufacture and maintain safe and effective devices. From a security context, these are also trustworthy and resilient devices. These devices can then be managed (e.g., installed, configured, updated, review of device logs) through the device design and associated labeling by the device manufacturers and/or users (e.g., patients, healthcare facilities). For health care facilities, these devices may also be managed within their own cybersecurity risk management frameworks, such as the National Institute of Standards and Technology Framework for Improving Critical Infrastructure Cybersecurity, generally referred to as the NIST Cybersecurity Framework or NIST CSF.
Security Risk Management
To fully account for cybersecurity risks in devices, the safety and security risks of each device should be assessed within the context of the larger system in which the device operates. In the context of cybersecurity, security risk management processes are critical because, given the evolving nature of cybersecurity threats and risks, no device is, or can be, completely secure. Security risk management should be part of a manufacturer’s quality system. Specifically, the QSR requires, among other things, that manufacturers’ processes address design (21 CFR 820.30), validation of the production processes (21 CFR 820.70), and corrective or preventive actions (21 CFR 820.100). These processes entail the technical, personnel, and management practices, among others, that manufacturers use to manage potential risks to their devices and ensure that their devices remain safe and effective, which includes security.
Specific security risk management documentation where FDA has recommendations regarding 354 their scope and/or content are discussed in the following subsections:
Threat Modeling
Third-Party Software Components
Security Assessment of Unresolved Anomalies
Security Risk Management Documentation
TPLC Security Risk Management
Security Architecture
Manufacturers are responsible for identifying cybersecurity risks in their devices and the systems in which they expect those devices to operate and implementing the appropriate controls to mitigate those risks. These risks may include those introduced by device reliance on hospital networks, cloud infrastructure, or “other functions” (as defined in FDA’s guidance “Multiple Function Device Products: Policy and Considerations), for example. FDA recommends that all medical devices provide and enforce the security objectives in Section IV, above, but recognizes that implementations to address the security objectives may vary.
Throughout this section, FDA outlines the recommended security controls and recommendations on how to document the resultant security architecture in premarket submissions through specific Security Architecture Views.
Cybersecurity Testing
As with other areas of product development, testing is used to demonstrate the effectiveness of control mitigations. While software development and cybersecurity are closely related disciplines, cybersecurity controls require testing beyond standard software verification and validation activities to demonstrate the effectiveness of the controls in a proper security context to therefore demonstrate that the device has a reasonable assurance of safety and effectiveness.
Security testing documentation and any associated reports or assessments should be submitted in the premarket submission. FDA recommends that the following types of testing, among others, be provided in the submission:
In order for users to manage security risks in devices, either by an end user or within a larger risk management framework like the NIST CSF, transparency is critical to ensure safe and effective use and integration of devices and systems. This transparency can be conveyed through both labeling and the establishment of vulnerability management plans. However, different types of users (e.g., manufacturers, servicers, patients, etc.) will have different abilities to take on a mitigation role, and the need for actions to ensure continued cybersecurity should be appropriate for the type of user.
In this section, FDA provides recommendations on:
Labeling Recommendations for Devices with Cybersecurity Risks
Vulnerability Management Plans
Vincent Balgos Afterward:
In general, cybersecurity continues to be a growing and significant issue in recent years. From the Experian data breach (2017), Marriot Hotel (2018), and LinkedIn (2021), the volume of sensitive data exposed at an alarming frequency poses a significant risk. In the healthcare sector, the Scripps Health cyberattack in my hometown of San Diego, CA, some estimate that the minimal impact is expected to be >$100 million. More significantly, the potential impact of sensitive patient data out in the public domain is immeasurable.
On the medical device front, the FDA has issued recalls on a variety of devices (insulin pumps, pacemakers, etc.) citing cybersecurity risks for years. As medical device technology advances, and interoperability becomes more common, the opportunity of cybersecurity risks is ever more present.
Medical companies have (or need to soon) started considering cybersecurity as part of their standard business practices. In addition to the suggestions mentioned in the FDA draft guidance, many of us at Jama Software have seen medical company initiatives that include hiring cybersecurity experts to proactively support these efforts, incorporating a dedicated cybersecurity focus within the overall risk management procedure, and/or continued monitoring of the products and systems for potential exploits.
Since a single vulnerability can have dramatic impact, a systems approach to cybersecurity can provide valuable perspective for various levels of mitigation. In complex systems, with multiple interfaces, data streams, etc., potential vulnerabilities increase at a non-linear rate.
But how does one measure the cybersecurity level of a product’s software? An emerging metric is the Common Vulnerability Score System (CVSS) that assesses software vulnerabilities in a quantitative value. Some organizations may include this CVSS as a design requirement during the product development phase.
This approach proactively incorporates cybersecurity best practices early in the design process, which in turn demonstrates security traceability into the design, and its subsequent testing. With Jama Connect, and specifically our unique ability to create Live Traceability™ across the product development process, this cybersecurity requirement can be continually monitored (along with all other requirements) to ensure that the final product is safe, effective, and secured from potential cyberattacks.
RELATED
One of the early steps I advise my clients to take when developing their medical device is to determine the class and classifications of their medical device. In conjunction with the complexity of the device, understanding the class and classification sets the foundation for your product development timeline and effort.
This post gives a basic introduction to FDA medical device classes and classifications and the implications for your product development schedule and requirements management.
What are FDA medical device classand classifications?
The FDA established three regulatory classes based on the level of control necessary to assure the safety and effectiveness of the device. Classification is based on the intended use of the device and indications for use, as well as the risk the device poses to patients and users.
There are three classes: Class I, Class II, and Class III. Class I devices are those with the lowest risk, Class II devices have a greater risk, and Class III includes devices with the greatest risk.
The FDA also established classifications for over 1,700 generic types of medical devices and grouped them into 16 panels, or medical specialties. Example panels include Cardiovascular Devices and Radiology Devices. Each of the generic types is assigned as Class I, Class II, or Class III.
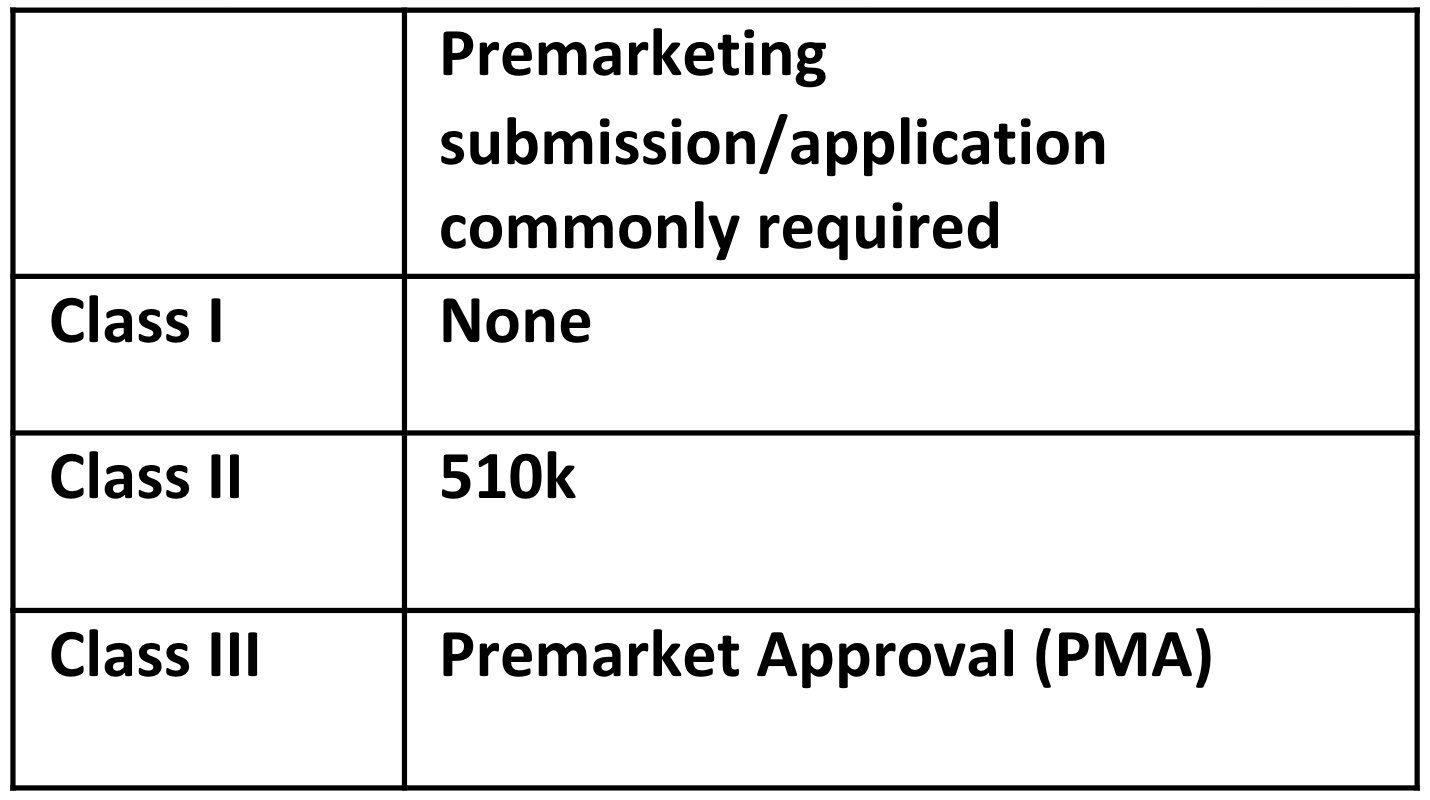
The class and classification of the device impacts what FDA premarket submission or application is required for clearance to market. The common premarketing submission or application for each class are:
Note: These are the common regulatory submission and applications for each class of device. There are exemptions, limitations on those exemptions, special controls that may apply, and exceptions, so be aware whether any of these applies to your device. For example, about a quarter of Class I devices are not exempt, and a 510k premarket submission is required.
As the process for the 510k submission is 30-90 days, and the process for the more in-depth PMA submission is 180 days to accept or reject, this time should be understood and planned into your product development schedule.
Similarly, expect elements from the required design control process and design history file to be included as part of a 510k and PMA. Also keep in mind that when design controls are required for your device classification, the full design history file can be scrutinized as part of an FDA inspection of your organization. Since the FDA evaluates whether a device is effective and ensures the risk to the patients and users is appropriately addressed, good requirements and risk management is key. It’s important to have an organized manner in which to demonstrate and document that risk management and user needs are successfully traced through design inputs, design verification, and design validation. A requirements management tool like Jama Connect™ allows for this traceability in an efficient, collaborative, and regulatory-compliant manner.
Understanding your device class and classification is a key step to understanding the path for FDA regulatory clearance and subsequent design control requirements for your medical device development. Knowing those expectations up front will make for a smoother medical device development journey.
Learn more about developing medical devices with Jama Connect!
In many ways 2021 was a continuation of the changes brought about in 2020, a year that’s been described as “unprecedented” and “unparalleled.” In a unique way, 2021 has offered us an idea of evolving innovations and technology on the horizon for teams across industries. These changing conditions will present a variety of new landscapes and will offer unique challenges, opportunities, and more than likely, many surprises.
As we enter a new year of further changes, Jama Software asked select thought leaders – both internal and external – across various industries for the trends and events they foresee unfolding over the next year and beyond.
In the second part of our five-part series, we ask Steven Meadows, Solutions Lead from Jama Software, and Ryan Moore and Carleda Wade, Solution Consultants from Jama Software – along with Thierry Marchal, Program Director for Healthcare Solutions at Ansys, and Ivan Ma, Medical Device Program Leadership, to weigh in on product and systems development trends they’re anticipating for medical device teams in 2022.
Read our other 2022 Industry Predictions here: Part One – Engineering Predictions,Part Three – Automotive Predictions, Part Four – Aerospace & Defense Predictions, and Part Five – Insurance Development Market Predictions.
Medical Device Predictions
Q: What product, systems, and software development trends are you expecting to take shape in 2022?
Steven Meadows, Jama Software:
Artificial Intelligence (AI)
AI, and in particular machine learning (a subset of AI), has been expanding rapidly over the past decade and now has a market of over $6.7 billion across med tech. I see this trend continuing with extra guidance from the FDA helping developers produce safer software with AI elements. AI is particularly exciting as it can enable streamlined MRI and CT scans, instant blood and at-home rapid testing, and a whole host of otherwise manual and often error prone activities.
Minimally invasive devices
Another trend I see in medical continuing in 2022 and beyond is the rapid growth and development of minimally invasive devices. Open heart surgeries are becoming a thing of the past, with cardiothoracic applications becoming the norm. Minimally invasive devices are also becoming heavily utilized throughout most orthopedic surgeries, and increasingly in urology procedures. We have seen a lot of start-ups attempting to bring the next best minimally invasive device to market.
Wearables
Medical wearable devices demand continues to rise, with apps on wearable devices beginning to provide recommendations to users, to improve their health. Heart rate and temperature sensing continue to be the most popular features on wearable tech today but there is increased investment in smart glasses, ‘earables’, and clothing.
Software development
Software will continue to be an integral part of many medical devices next year and beyond. We consistently see that our software customers have adopted, or are transitioning to, an Agile methodology when creating their medical system, a shift that has been happening for some time. Gone are the days of slow waterfall-based development practices.
As I mentioned before, AI based software development will continue to trend and grow next year, and AI software development practices will be geared around a tailored regulatory framework, good machine learning practices, and a patient centered approach. The FDA will continue to refine guidance around AI/Machine Learning (ML.)
Cybersecurity and in particular data security will continue to be a top priority for our software customers, with data breaches and hacks on the rise. The FDA is planning to soon release extra guidance around cybersecurity in medical devices with a particular emphasis on quality system considerations and content of premarket submissions.
Ryan Moore, Jama Software:
I anticipate a trend toward more software-basedtoolsets and digitization. Also, an increased amount of automation and robotics.
Carleda Wade, Jama Software:
I expect greater integration of software tools such as with Requirements Management software with eQMS software.
Thierry Marchal, Ansys:
The dramatic COVID pandemic has amplified a trend that appeared a decade ago – progressively calling for the adoption and deployment of in silico medicine. Stormed with the COVID pandemic, the world could not wait for 10 to 15 years to get a new vaccine fully tested and approved using a traditional approach. Furthermore, this COVID disease is impacting people differently, while elderly people are impacted the most, possibly leading to long term disabilities and treatments.
EDITOR’S NOTE: According to The University of Sheffield’s Insigneo Institute, In silico medicine (also known as ‘computational medicine’) indicates modeling and simulation technologies that directly contribute to the prevention, diagnosis, prognosis, treatment planning & execution, or management of the disease. In silico methods complement traditional in vivo approaches (working with animals and human beings) and in vitro testing (working in a lab.)
Thierry Marchal, Ansys:
This pandemic highlights what we have been observing for decades: with an aging population, the cost of chronic diseases (especially for old people) is weighing a lot on the healthcare cost. The solution will come from personalized medicine and preventive medicine combining pathologies detection in its early stage to treat diseases before they impact people. The continuous monitoring of patients to detect pathologies early are calling for e-health and mobile health (m-health): wearable and implantable devices continuously and safely watching the patient and soon feeding your own digital twin or Personal Digital Avatar (a computer model of yourself, connected with you, and properly stored in the cloud to guarantee your data privacy) is now an emerging trend. Digital twins will be a crucial innovation for the software necessary to use patient specific data to predict the evolution of the connected patients.
As medical innovation will be essential soon, this evolution cannot be slowed down by an extremely long and costly regulatory approval process: a digitalization of drug and medical device approval, including in silico (clinical) trial is another major trend that we are observing.
Ivan Ma, Medical Device Program Lead:
Technologies that enable capabilities via telemedicine with more than just a face and a voice over the screen will make “seeing” the doctor digitally as common place as working from home.
Protecting patients, physicians, and staff by reducing or eliminating exposure to harmful forms of imaging such as fluoroscopy will always be a valuable endeavor.
Q: In terms of product and systems development, what do you think will remain the same over the next decade? What will change?
Steven Meadows, Jama Software:
A lot of the areas I mentioned that will trend in 2022, will more than likely trend over the next decade.
AI, not only in medical but across most industries, is on the rise and patients will continue to see improved outcomes, quicker diagnoses, and a better quality of life.
Medical wearable technology will continue to evolve, with improved functionality helping keep us fitter and safer.
Minimally invasive devices will continue to be expanded across different medical areas, improving recovery times, and surgery outcomes. Increasing emphasis on cybersecurity will resume, to prevent malicious actors from hacking sensitive data and for connected systems to remain operational.
COVID-19 is not going away any time soon so a reliance on collaborative product development tools, like Jama Software, will continue to be an integral part of any organization placing an importance on quality and overall product and patient outcomes.
Ryan Moore, Jama Software:
Medical devices will still be comprised of mainly hardware (HW)/software (SW), while automation and robotics will be a driving force.And users will still need to power devices.
Carleda Wade, Jama Software:
Medical device development will mostly stay the same as the regulations are fairly stable.However, I do expect increased usage of software in the process since much of the workforce may now be working in various locations. I also expect to see an increase in AI and Software as a Medical Device (SaMD)as a whole in the industry.
Q: What are some of the biggest challenges you think engineering firms will be working to overcome in 2022?
Thierry Marchal, Ansys:
Contrary to other industries, innovation in healthcare faces the challenge of human variability: as we are all different, it is not acceptable that a treatment would work well for a few people and poorly for most others. Using computer modeling and simulation (CM&S a.k.a. in silico methods) is a cost-effective way to test new treatments efficiently without compromising with patient safety, on large cohorts of virtual patients.
An accurate prediction of the treatment outcome for a given patient will require combining traditional modeling techniques with biological models, often extracted from big data observations using AI.
Finally, the community remains skeptical about the reliability of computer model and digital evidence to predict correctly and accurately what is happening in the real life. This credibility challenge will be continuously addressed by more validations and comparisons with in vitro and in vivo data.
Ivan Ma, Experienced Medical Device Developer:
The development of medical device hardware will always require teams to gather in front of and handle hardware. As the pandemic continues, and in-person work still not back to what it once was, it is time to think of strategies that maximize time on developing hardware with minimal people in the room. Could digital Operating Room technologies be reconfigured and brought in to support the verification and validation phases of medical device development? The tenets are the same. Minimize the number of people in the room, allow the expert to drive the hardware from afar, make data acquisition, observation and learning access as easy as clicking on a link.
Q: How do you foresee regulations shifting in medical device product and systems development over the next decade?
Steven Meadows, Jama Software:
Although AI has been utilized in medical device and life science products for decades, guidance has been lagging. It’s clear to see that AI has incredible benefits for patients, and so there will continue to be increased regulatory guidance available, to help developers build products which contain AI in a safer and standardized manner. Check out a blog Jama Software authored around machine learning in SaMD and shifting regulations here.
The medical device regulation (MDR), which was accepted and implemented in the EU in 2017, has been amended over the past few years. The latest change focuses on increasing the responsibility and accountability of medical device companies throughout the entire development of a product. Expect more guidance over the next decade, as gaps are addressed to ensure product safety is at the forefront of any market clearance.
Ryan Moore, Jama Software:
I would expect compliance and regulations to be more defined as we move forward. Currently, there is a lot of grey area in how standards/regulations are interpreted. The FDA will need clearer guidance for modern technology that arises
Carleda Wade, Jama Software:
With an uptick in AI and SaMD I expect the FDA and foreign regulatory bodies to begin providing more clarity on how these complex systems and softwareneeds to be developed, validated, and maintained.
Q: What changing regulatory guidelines do you anticipate having an impact on companies in 2022?
Thierry Marchal, Ansys:
The US authorities developed, without any doubt, the most advanced regulation in terms of adoption of computer model results for the regulatory approval process. However, the number of published cases reporting the actual use of in silico methods by sponsors remains limited. New results will be published in 2022 further encouraging companies to confidently follow this process.
The European Medical Device Regulation (MDR), in application in Europe since May 2021, is opening the door to digital evidence and in silico approach; unfortunately, the process to validate a model and report simulation results has not been clarified yet. Similarly, as the European authorities are updating their pharmaceutical strategy in 2022, this document is expected to make references to computer modeling and simulation.
In the rest of the world, we observe a growing interest for in silico methods and the need to regulate this approach. After years of compiling information and experience from other parts of the world, we are expecting that some Asian authorities will start to communicate about this topic in 2022.
Q: What sorts of process adjustments do you think medical device development teams will need to make to be successful in 2022?
Steven Meadows, Jama Software:
One of the biggest process issues we see across our customers is that they view quality as a secondary function and a necessary checkbox activity once a product is developed, or close to being finalized. Because of that, products tend to contain more defects, resulting in more in field CAPAs, negative patient outcomes and even have a greater chance of being recalled. Development teams should ensure quality is prioritized from the get-go.
Although we have noticed a large shift with software teams adopting an Agile approach when it comes to development, we can’t emphasize enough the importance of adopting a lean and iterative approach.
Ryan Moore, Jama Software:
Continue to follow standardized process with focus on aligning with FDA / compliance in parallel with building safe products
Carleda Wade, Jama Software:
Teams will need to find the sweet spot of trying to collaborate and innovate while not being physically in the same location at the same time. Due to the pandemic, many teams will now transition to being fully remote permanently or have a hybrid schedule and it may make things more difficult when developing new products with a cross-functional team.
Q: From an engineering toolset perspective, what are some of the processes you believe forward-thinking firms will be working to leverage or incorporate into their process and why?
Theirry Marchal, Ansys:
As we see the healthcare community adopting in silico method with more enthusiasm both for design and regulatory approval (not mentioning emerging clinical applications), the software community is rushing to deliver the supporting in silico tools. We could mention three major avenues followed by Ansys.
As in silico clinical trials requires a large number of simulations, specific Simulation Process and Data Management (SPDM) tools structuring the digital evidence following the ASME VVUQ40 standard will greatly facilitate the adoption of this approach. Ansys is customizing its Minerva tool in a Minerva VVUQ 40 template for medical device and pharmaceutical companies.
It is crucial to model the behavior of a new treatment in its working environment, usually the human body. Human organs such as the heart, the lungs, and the brain are extremely complex to model and validate: this requires a collaboration of various actors. Ansys is closely working with leading academics, global healthcare companies and startups, and clinicians to continuously update and validate virtual hearts, lungs, and brains in collaboration with a large ecosystem.
Although everybody recognizes the potential of digital twin and Personal Digital Avatar, the necessity of developing quasi-instantaneous modeling capabilities despite the complexity of the model is a real challenge. Ansys is taking advantage of its experience with other industries already using digital twin of equipment to help the pharmaceutical industry to develop digital twin of drug manufacturing equipment and initiate the first steps towards Personal Digital Avatars.
Ivan Ma, Medical Device Program Lead:
Engineers need tools that minimize the volatility and error that occurs when bringing hardware and electronics from the drawing board to parts in hand. How do organizations reduce hardware prototype cycles, recognizing that even if the design is right on paper, the production of that design can pass through many hands before it is made into a real part, thus decreasing the probability of a successful build.
Q: What do you think will be some of the differentiators between a company surviving to see 2030, and those that do not?
Steven Meadows, Jama Software:
Along the lines of what I mentioned companies should focus on in 2022 to be successful, medical device companies that embrace a proactive approach to quality(Link here – https://www.jamasoftware.com/blog/three-ways-to-proactively-vs-reactively-incorporate-design-controls-in-medical-device-product-development/ )will ultimately find fewer issues with their products, improve customer satisfaction, and stay competitive for the foreseeable future. Companies that invest in best-of-breed medical device development solutions like Jama Connect will have the upper hand in reducing risk and complying with various standards and regulations.
New cutting-edge technologies in the medical field – like robotics nanotechnology, AI, and wearable health tech devices – bring complexities for medical device companies and risk for patients and consumers. Jama Software will continue to serve as the leading solution and partner to help innovative companies bring medical products to market, in a collaborative, safe, and efficient way.
Defined process, building quality products that meet a specific market need
Carleda Wade, Jama Software:
Truly understanding the market needs and being willing to allocate resources so that you become a pioneer in a certain industry will help a lot of companies to succeed; however, having passionate and qualified personnel will be the biggest success factor. Companies that only want to develop “me too” devices will struggle to gain market share, and even if they are able to survive it will be difficult for them to thrive.
Q: Where do you see Jama Software fitting in as the product development landscape evolves, and what can our customers expect as 2022 approaches?
Steven Meadows, Jama Software:
Jama Software provides incredible value for hundreds of medical device companies ranging from start-ups to established corporations. With COVID-19, we’ve seen an uptick in customers looking for an easy-to-use product development tool that offers differentiating collaborative capabilities to keep people connected throughout different design stages. Jama Software continues to invest heavily in the application, and we will see improved core product capabilities as well as integrations, strengthening the tool’s ability to seamlessly work alongside other systems.
Ryan Moore, Jama Software:
Jama Connect can be used as the core toolset for requirements, testing, and review/approvals. Jama Software’s ability to branch out into further workstreams (in order of priority: risk management, test automation, modeling/design outputs) will bring exponential value to teams using Jama Connect.
Carleda Wade, Jama Software:
I can see Jama Software bringing value to not only the start-up but the established company in the future. As a medical device company, the first goal of the company is to make a good product that meets a market need, this happens even before establishing a quality management system in many cases. Having a tool like Jama Connect will ensure that as products are developed that all the bases are covered. Later, when it is time for regulatory approval having a tool like Jama Connect in place will make the regulatory submission process very simple.
Thanks for tuning into our 2022 Predictions Series! To see some of the incredible products, software, and systems our customers are building with Jama Connect, visit our CUSTOMER STORIES PAGE.
Complying with FDA Design Control Requirements Using Requirements Management Principles
Mercedes Massana: So agenda for today is we’re going to talk a little bit about the design controls and what they are. We’re going to talk about requirements management and what that is. And then we’re going to talk about how the two relate to each other. Then we’ll discuss how we can build a good requirements management process and how that process can help us improve compliance to design controls.
Okay. So let’s get started. All right. So by the end of this presentation, hopefully you’ll have a good general working knowledge of design controls. You’re going to understand requirements management and how to evaluate and accept requirements, how to perform bi-directional traceability, how to use requirements attributes to help manage the requirements, and how requirements metrics can help you make better decisions. We’re going to learn how to manage and control requirements changes, and then how to use attributes, especially a requirements credit quality attribute to help us on manage and identify essential requirements and requirements that are critical to quality.
FDA Design Controls
Mercedes Massana: So let’s start with design control background. So design controls have been part of the FDA QSR since 1997. So it’s been a long time. The FDA initially implemented design controls to try to help medical device manufacturers identify deficiencies with design input requirements, to identify discrepancies between proposed designs and requirements, and increase the likelihood that the design transferred to production is going to translate into a device that is appropriate for the intended use and satisfies the user needs. And they did this in relation to having a lot of issues in the field with medical devices. So they thought design controls would improve the quality of the medical devices that were being approved.
In the FDA design control guidance, they state that developing a solid foundation of requirements is the single most important design control activities. So that kind of tells you how important FDA thinks design control requirements management is. So design controls is made up of several elements. There is design and development planning, design inputs, design outputs, design review, design verification, design validation, design transfer, design changes, and design history file.
Mercedes Massana: Now, most of those, even though requirements really falls under design inputs, and that’s what everybody understands by design inputs, most of these items relate to requirements in one way or another. So design outputs, we have to develop design outputs that will conform to design inputs. For design reviews, we’re going to review our design inputs to make sure that they’re appropriate and not conflicting or unambiguous. Design verification will confirm that design outputs meet the design input requirements. And design validation will confirm that the user needs and intended uses are met, which is their another form of design input.
And then design changes, obviously managing requirements changes is important. And obviously all our design inputs or all of our user needs documents or system requirements or product requirements documents will end up in the design history file. And then obviously our deliverables that user needs that we create, or system requirements, product requirements, all become part of our design history file.
Requirements Management
Mercedes Massana: So let’s talk about requirements management. So what is requirements? Why do we need requirements management? Why would we want to do this? Well, if your projects are late because you’re trying to introduce changes at the end, or if sometimes your developers can really figure out what the requirements may mean and they’re making their own interpretation, those are all issues that can be addressed through requirements management. In fact, it is said that approximately for IT projects, which encompass software obviously, that at least 50% of projects are late, or delivered with reduced functionality or over budget.
Mercedes Massana: If you have a good requirements management process, this will improve those statistics and make projects more successful. So what is a requirement? So for IEEE, a requirement is a property that a product must have to provide value to a stakeholder. That’s the IEEE definition. The FDA definition is a condition or capability needed by a user to solve a problem or achieve an objective. So couple of key items here is, something that is needed by somebody, right? So that tells you that anything that goes into your requirements has to have a purpose. Why is it there? And it’s needed to solve a problem. So it has to solve a problem for the user, right? So if it’s something that a software developer thinks is a cool feature, but nobody needs it, then it really shouldn’t be a requirement.
So requirements management process has four main goals. It tries to ensure that there is an understanding of requirements, but all the stakeholders, that they commit to those requirements or approve or agree that those are the requirements that need to be implemented. That we manage requirements changes. And we’ll talk what managing requirements changes means a little later. And then we identify inconsistencies between project work and requirements. And what that means is that you can’t have a project plan that says you’re going to be done in a year if you have 10,000 requirements to implement, right? So you have to have consistency between the requirements and the other deliverables of the project.
So to obtain an understanding of the requirements, first, we need to know who are we solving the problem for? Right? So we need to determine how we identify the stakeholders. So who are those requirements providers, right? Those are the stakeholders. We need to know how we’re going to evaluate the requirements, how we’re going to know if the requirements are good and complete. And then we need to know who needs to approve the requirements and when do requirements get approved. So that’s how we obtain an understanding of requirements.
Then a commitment to requirements is basically that agreement between the different stakeholders that says, if you develop these set of requirements, we’re going to have satisfied customers, that our agreement is usually made through approval of the requirements.
Managing requirements changes is, obviously the one constant in life is change. So requirements will also change. But it’s very important to manage changes so that the rest of the product reflects the impact of those changes, right? Nothing comes for free, and requirements changes usually are either going to cost more, and we’re going to have to add more resources or we’re going to have to add more scheduled, but something will have to be done in order to manage the impact of that change.
Requirements Traceability
Mercedes Massana: And then maintaining bidirectional traceability. This requires that requirements be traced forward from higher level requirements to lower level requirements and that every lower level requirement be able to be traced backwards to its parent or a higher level requirement. Additionally, other elements like design, tests and risks should also be included in our traceability. And later on, I’ll show you a fully elaborated trace matrix that shows the relationship between pretty much every deliverable that we’ll create as part of our development process.
All right. So what’s the connection between requirements management and design controls? So this table will show us a little bit of that. So these requirements management elements relate to design controls as follows. So design inputs are defined as the physical and performance requirements of the device that are used as the basis for device design. So the FDA tells us that design inputs must be appropriate. That we need to address incomplete, ambiguous or conflicting requirements. And that these requirements related to design inputs are addressed by the requirements management element and obtaining an understanding of requirements.
Design outputs are defined as the results of the design effort at each design phase and at the end of the total design effort. The FDA says that design controls must be able to evaluate the design outputs conformed to design inputs, that we must be able to establish acceptance criteria so that we can verify the design outputs conform to the design inputs. And then we identify the essential requirements or essential to proper function requirements. So again, requirements management can help us meet this part of the regulation.
FDA design controls also state that we must identify document, verify, validate and approve design changes before implementation. Additionally, the design control guidance specifies that the trace matrix is a form of verification. So both of these areas are also covered by requirements management. And here, we can see that there is definitely a relationship between FDA design controls and any requirements management process.
Design Controls have been an FDA Quality System Regulation since 1997. Having worked on developing products in the regulated medical device industry for over 35 years, I have compiled a list of the five key takeaways for implementing design controls and achieving success in commercializing medical devices:
Design Controls not only help achieve regulatory compliance, they help develop better products
Design Inputs lay the foundation for product development, building a good foundation
Don’t underestimate the power of Bidirectional Traceability
Know the difference between Verification and Validation
Risk Management is a vital part of Design Controls
#1 – Design Controls not only help achieve regulatory compliance, they help develop better products
Many companies think that design controls are a burden to development organizations imposed by the FDA, and it’s the price to pay for playing in the medical device field. However, what is often overlooked is that design controls only define the basic minimum requirements necessary to develop a product that can…
…meet the needs of the user.
…be designed to be safe and effective.
…be reliably manufactured.
…be verified and validated.
…maintained and updated throughout the product lifecycle.
These are all things that any development organization should do to successfully deliver products to market. I like to say that if you are doing the right things in product development, compliance comes for free!
#2 Design Inputs lay the foundation for product development, building a good foundation
The FDA defines design inputs as the physical and performance requirements of a device
that are used as a basis for device design. To generate adequate design inputs, the foundation upon which product development is built, the user needs must first be well understood. These needs, ideally written with the voice of the user, must then be translated into design requirements. In contrast to the user needs, these design requirements should be written using the voice of the engineer, and as such should be measurable and testable. Furthermore, design requirements should be traceable to a specific user need, risk control, or standard that necessitates the existence of said design requirement.
Research has shown that, on average, companies that are successful at developing products spend about 25% of the product development time on the generation of user needs and the subsequent design requirements. The return on this investment of time and resources reduces the need for rework and redesign, and ultimately leads to higher customer satisfaction. Failing to make the investment ensures that design inputs are complete and correct is analogous to building a house on quicksand, where the flaws in the foundation can cause issues throughout the construction and subsequent (likely short) lifetime of the house. Issues with requirements will impact development, verification, validation, and user acceptance of the product, so spending the time to get requirements right will be well worth the effort.
#3 Don’t underestimate the power of Bidirectional Traceability
In an audit, the trace matrix should be valued as a friend! Having and maintaining bi-directional traceability throughout the product lifecycle provides a number of benefits:
Effecting project tracking
Thorough change impact analysis
Ease of making future changes
Re-use of elements of the design
More effective issue resolution
To derive these benefits, the relationship between the following entities should be established:
User Needs and Design Requirements
User Needs and Validation
Design Requirements and lower-level requirements
Design Requirements and Verification
Lower-level requirements and verification
Lower-level requirements and Design Outputs
Risk Controls and Design Requirements
In creating a trace matrix that has views to show all the bi-directional relationships of each of the elements described above can help answer most questions from an auditor. With this level of traceability, I can trace from a user need all the way through implementation and test.
#4 Know the difference between Verification and Validation
The terms “Verification” and “Validation” often get combined and abbreviated to V&V; however, these activities are vastly different.
Verification is confirmation by examination and provision of objective evidence that specified requirements have been fulfilled. It is design-centric and answers the question “Did I build the product right?” Verification also entails gathering objective evidence that the design behaves as intended through the use of observation (visual inspection), measurement (values and tolerances), testing (function) or analysis (reviews).
Validation is confirmation by examination and provision of objective evidence that the particular requirements for a specific intended use can be consistently fulfilled. Unlike Verification, this is a user-centric term, and answers the questions “Did I build the right product?” and “For whom is this the right product?” Validation entails gathering objective evidence that the design satisfies the user needs through the use of Usability Studies/Human Factors Studies, Clinical Evaluation/Clinical Studies, Customer Surveys, and through Analysis of Verification Data.
Knowing the difference between Verification and Validation is of quintessential importance for ensuring customer satisfaction and regulatory acceptance of the product.
#5 Risk Management is a vital part of Design Controls
The elements of design controls are Planning, Design Inputs, Design Outputs, Design Reviews, Design Verification, Design Validation, Design Changes and the Design History File. So, what happened to Risk Management? Risk is mentioned in Design Control Regulation (QSR 820.30) all of one time, under Design Validation. The statement simply reads “Design validation shall include software validation and risk analysis, where appropriate.”
Fortunately, the FDA Design Control Guidance elaborates on requirements for risk management. The guidance includes this paragraph:
Risk management begins with the development of the design input requirements. As the design evolves, new risks may become evident. To systematically identify and, when necessary, reduce these risks, the risk management process is integrated into the design
process. In this way, unacceptable risks can be identified and managed earlier in the
design process when changes are easier to make and less costly.
The takeaway from this is that although risk management is just cursively mentioned in the QSR Design Control regulation, the intent of the regulation is that Risk Management be practiced starting from the point where design inputs are known and practiced throughout the product life cycle. You cannot be compliant to the design control regulation without having an adequate risk management file.
Conclusion:
Design Control regulations have been around since 1997, but many manufacturers still have problems complying with design controls. Focusing on the best practices outlined above will derive the most benefit from implementing Design Controls, will lead to a more predictable development cycle, and ultimately result in higher-quality products that can be enhanced and maintained throughout their lifecycle.
This post on Software as a Medical Device (SaMD) development is written by Mercedes Massana, the Principal Consultant of MDM Engineering Consultants.
SaMD software is software intended to be used for one or more medical purposes that do not require embedding in a medical device. These purposes can range anywhere from helping to diagnose or treat disease, helping in the clinical management of a disease, or providing information to help with the clinical management of a disease. SaMD differs from other medical device software in that it will operate on different platforms and will interconnect with other devices, carrying with it an increased cybersecurity risk and a commensurate increase in the use of off-the-shelf software.
On the surface it may appear that the development of SaMD software is no more difficult than the development of Medical Device embedded software, but appearances can be deceiving, and the development of SaMD products can be quite challenging. In order to deal with these challenges, there are four key best practices that should be followed for SaMD software development.
These practices are:
Make use of standards and guidance documents
Apply the right level of rigor
Understand the difference between Verification and Validation
Implement a post-market monitoring program
Best Practice #1– Making Use of Standards and Guidance Documents
Although standards development organizations and regulatory bodies have only started to scratch the surface in the creation of standards and guidance documents to help SaMD development organizations, there is a sufficiently detailed body of work available to help development organizations succeed. The most relevant of these are the documents generated by the International Medical Device Regulators Forum (IMDRF) related to SaMD and IEC 82304 Safety and Security of Health Software Products. The IEC standard points to other well-known standards, such as IEC 62304 Software Development Lifecycle, IEC 62336 Usability Engineering and ISO 14971. Additionally, several FDA guidance documents are available that apply to all medical device software and are useful for the development of SaMD, these include General Principles of Software Validation, Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices, Off-The-Shelf Software Use in Medical Devices and the FDA pre and post market cybersecurity guidance, as well as other guidance documents
Best Practice #2 –Applying the Right Level of Rigor
Within the development of SaMD, a clear understanding of the scope and intended use of the product is necessary, and to that end, it is necessary to have a method to gauge the risks associated with SaMD use. The IMDRF “Software as a Medical Device” Possible Framework for Risk Categorization and Corresponding Considerations, IEC 62304 , and FDA Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices all provide a method for risk based classification of SaMD. The rigor applied to the development of SaMD should be commensurate with the level of risk. IEC 62304 uses the Safety Classification to define the level of rigor of activities to be performed in the software lifecycle based on the level of risk. Ideally, the development process is sufficiently flexible to avoid the over-engineering or under-engineering of the SaMD in question. A development process that is adaptable requires organizational maturity and experience, in order to perform the right set of activities and understand the value provided by those activities.
Best Practice #3 – Understanding the Differences Between Verification and Validation
SaMD is a medical product, a system in and of itself. Therefore, SaMD system requirements must be verified to have been implemented correctly, and SaMD user’s needs must be validated to ensure that the product satisfies the user’s need and that the right product was built for the customer. Validation typically includes human factors testing, clinical evaluation to determine the efficacy of the software for its intended use, and a demonstration that risk controls are effective. This requires more than just your standard set of software testing, which typically consists of code reviews, unit testing, static analysis, integration testing and requirements-based testing.
Best Practice #4 – You Are Not Done When the Software is Released
Your SaMD software has successfully completed verification and validation, has been cleared by regulatory agencies, and is now ready to go live. Time to breathe a sigh of relief and congratulate the team for a job well done. Pop the champagne and throw a party, you have all earned it. Enjoy the festivities, but afterwards turn your attention promptly to monitoring the performance of the SaMD post launch. Rigorous and exhaustive as your testing was, you can never anticipate every possibility. Be ready to respond to identified software defects and emerging cyber threats and deploy patches or software fixes as issues are identified. The nature of cybersecurity risks is ever-changing, and no system, regardless of how well-designed, or rigorously-tested, is free of software defects. Additionally, customer input can provide opportunities for enhancements and improvement. However, be aware that proposed changes can impact the intended use of the device, the classification of the SaMD, and can ultimately result in additional regulatory submissions. It is paramount that prospective SaMD developers evaluate changes thoroughly and plan actions related to these changes to avoid surprises that can delay, or outright derail, SaMD implementation.
In summary, SaMD can provide great benefits to the user; however, to successfully launch SaMD that meets the user’s needs, several best practices should be utilized in their development. These best practices, making use of standards and guidance documents, applying the appropriate level of rigor in the development of SaMD, understanding the difference between Verification and Validation, managing change appropriately, and implementing a good post-market monitoring program. These best practices are indispensable in ensuring a safe and effective SaMD product.
So, what is this Safety and Performance Based Pathway? Why did the FDA create it? How might it affect medical device development? Let’s examine those questions.
What is the Safety and Performance Based Pathway?
The existing Abbreviated 510(k) Program, established in 1998, was designed to lower the regulatory burden on both manufactures and the FDA and speed new medical devices to market. It permits manufactures to gain FDA approval by demonstrating their new device provides equal or better performance than an approved device that’s currently on the market (a so-called, “predicate device”).
The new Safety and Performance Based Pathway will allow manufacturers to gain approval by demonstrating that their new device meets a set of FDA-identified safety and performance criteria for its device category, rather than making a direct comparison with a predicate device.
The scope of this new pathway is limited. Issued on February 1, 2019, the guidance document states that this pathway applies only to specific device types the FDA understands very well and for which it has issued safety and performance criteria.
To that end, the FDA has begun releasing and soliciting industry feedback on guidance for specific device categories, starting with four documents issued on September 19, 2019. In each document, the FDA identifies the intended use and design characteristics of the device class along with the testing performance criteria for the class. The four device types for which the FDA has issued guidance are:
Conventional Foley catheters
Cutaneous electrodes for reporting purposes
Orthopedic, non-spinal, metallic bone screws
Spinal plating systems
The FDA plans to expand the program by rolling out many more such documents in the coming months.
Why has the FDA created the Safety and Performance Pathway?
The new pathway is part of the FDA’s effort to reduce the regulatory burden on both medical device manufacturers and the agency while speeding safer, more effective products to market with FDA approval. With the Safety and Performance Pathway, the FDA hopes to reduce manufacturer’s dependence on direct comparison with predicate devices, especially older predicates that rely on dated technologies. According to the agency, of the 3,173 devices cleared through the Abbreviated 510(k) pathway in 2017, roughly 20% were predicated on devices that were more than 10 years old.
When the new pathway was first announced in November 2018, FDA Commissioner Dr. Scott Gottlieb and Dr. Jeff Shuren, director of the FDA’s Center for Devices and Radiological Health (CDRH) said in a press release that, “Rather than looking to the past as a baseline for safety and effectiveness — and rely on predicate devices that are sometimes decades old as our point of comparison — our premarket review would be based on a contemporary baseline that looks to the future and a baseline that can be updated as technologies advance.
“Through this new path, a company would demonstrate that a novel device meets modern performance-based criteria that have been established or recognized by the FDA and reflect current technological principles,” they wrote. “These criteria would reflect the safety and performance of modern predicate devices. We’d like this efficient new pathway to eventually supplant the practice of manufacturers comparing their new device technologically to a specific, and sometimes old, predicate device.”
The press release also stated that the agency is investigating whether it can retire older devices as eligible predicates. Such a move could require Congressional approval, however.
“To be clear, we don’t believe devices that rely on old predicates are unsafe, or that older devices need to be removed from the market,” Gottlieb and Shuren wrote in their statement. “However, we believe that encouraging product developers to use more modern predicates would give patients and their doctors a choice among older and newer versions of a type of device, promote greater competition to adopt modern features that improve safety and performance, and help make sure that newer devices reflect more modern technology and standards that can improve patient care and outcomes. It would help the overall product environment continue to evolve in the direction toward more modern performance standards.”
Learn how Jama Software can help medical device developers better manage risk with ISO 14971 by downloading our white paper.
Pushback from Medical Device Industry Lawyers
Some think that with their stated goal of encouraging innovation, the FDA is overstepping its authority.
In an email to MobiHealthNews, Bradley Merrill Thompson, a lawyer who counsels on FDA regulatory issues, said phasing out old devices would be “essentially forcing companies to innovate regardless of whether the marketplace perceives a need,” thus driving unnecessary R&D and higher healthcare costs. He believes that if the FDA feels it needs to drive innovation through new regulatory changes, it would need to convince Congress to update the Medical Device Regulation Act of 1976.
“The 1976 statute is very clear, and FDA can’t simply ignore it,” Thompson wrote. “If FDA wants to change the statute, then to Congress they should go. Congress in turn could bring together all of the various stakeholders including patients, healthcare professionals and medical device manufacturers to talk about what they need and the best way to do this. FDA has no mandate to do this on its own.”
Possible Effects on Medical Device Development
Thompson’s legal concerns seem to apply only to the FDA’s investigation into retiring older predicate devices from the original Abbreviated 510(k) Program, however. From an engineering standpoint, the new Safety and Performance Based Pathway appears to offer manufacturers some important benefits.
First, the alternative pathway will allow manufactures to focus on surpassing safety and performance criteria rather than on making a comparison with a predicate device. Designers will have greater liberty to move away from predicate designs and technologies, since there is no need to make a direct comparison with a predicate device.
Second, for new devices in the FDA’s “well-understood” categories, the new pathway should speed time to market. The impact will be limited initially, but it should increase as guidance for more device types is released.
Finally, by establishing modern performance benchmarks for FDA approval that can be updated over time, the new pathway should help increase the safety and performance of new medical devices, at least those in designated device categories.
As we look to the future of medical device development, we can expect to see additional and extended guidelines from the FDA and other governing bodies on how to safely bring new products to market in a way that enables innovation. Stay tuned.
Based in Minnesota, Elucent Medical is a medical device developer focused on creating precise, wireless surgical navigation tools for tumor identification and excision. Founded in 2014, Elucent is helmed by three veterans of venture-backed biotech companies, CEO Laura King, CTO Dan van der Weide, and Dr. Fred Lee; as well as Dr. Lee Gravatt Wilke, Director of the University of Wisconsin Health Breast Center and chair of the research committee of the American Society of Breast Surgeons.
The team at Elucent has been using Jama Connect™ for about a year to manage requirements, tests, and reviews throughout the product development cycle.
Jason Hiltner, Elucent’s VP of Research and Development, had worked with requirements management solutions like CaliberRM before, and he had seen other product teams depend on IBM Rational DOORS and Microsoft Word and Excel. With those experiences in mind, he knew he needed a more intuitive, centralized platform to support innovation at Elucent.
Jama Connect makes Elucent’s product development process more efficient and more collaborative, while facilitating traceability from requirements through testing in compliance with FDA regulations.
And Elucent is creating some amazing things with the help of Jama Connect. Since it’s difficult to locate precise spots in the body, especially in soft tissue, Elucent has developed the SmartClip™ — a tiny implantable product that marks the location of cancerous tissue so surgeons can identify and excise the cancerous tissue more easily. Patients don’t have to endure repeated excisions because surgeons haven’t located or removed all of the cancerous tissue, and the SmartClip™ eliminates the need for painful and costly radiology localization.
The EnVisio™ Navigation System, which is designed to enable real-time 360° navigation in the surgical sightline and provides distance, depth and direction to the SmartClip(s): guiding a surgeon’s tools to make tissue excision and removal even more exact. The team’s hard work was rewarded when the EnVisio™ Navigation System was granted FDA clearance in 2019.
Elucent’s first market will be surgical excision in breast-conserving surgery, and their technology has wide applicability for localization of cancerous lesions throughout the body. Hiltner says the initial response to the clinical product has been “phenomenal for sure.”
We asked Hiltner and Judson Guericke, Senior Director of Quality at Elucent, to tell us more about how they’re leveraging Jama.
Jama Software: What corporate or industry challenge led to your decision to seek out a solution and ultimately purchase Jama Connect? Was there one particular problem that became a huge pain point?
Jason Hiltner: Obviously, in our industry, we have to have well-documented requirements that start with the customer requirements, go to the product requirements, link to the design descriptions and the software requirements, and lead to verification. If you’re going to do that, you need to start with something that allows you to have traceability. We also needed something with a central database, so we weren’t managing a bunch of different revisions of documents and emailing documents again. The big thing I wanted was a platform I could very easily maintain and not have questions about: I would know where things were and be able to easily do reviews. I also knew we needed something stable, robust and cloud-accessible.
How long have you been using Jama Connect, and what was the adoption process like?
Hiltner: We’ve been using Jama Connect for about a year. We’re a startup, so I talked over the idea [of a requirements management solution] with a small group of people. We’re a very lean and focused organization; we’re at only 11 people. We compared a couple of tools and got recommendations from people who had done a more exhaustive search. We went with Jama Connect in spite of the fact that it was more costly than some other tools. The deployment and implementation process was as smooth as I could have hoped for. We had a bunch of sessions with Zeb Geary, a Jama Professional Services consultant, and we were up and running, creating requirements in less than a week. It’s as flexible as I hoped it would be.
“We were up and running, creating requirements in less than a week.” – Jason Hiltner, VP of Research and Development at Elucent Medical
What was the feature/capability/business aspect of Jama Software that won you over?
Guericke: The collaboration and customer support for implementation. The biggest benefit for us is the efficiency in completing and gaining agreement on requirements and testing procedures.
Hiltner: We wanted something that would be very usable, something that would be maintained and around for a while. Our product has customer requirements that flow down to product requirements, which flow down to software requirements and design descriptions. We also maintain our verification suite within Jama.
Guericke: We needed a solution that would facilitate traceability from requirements through testing to comply with FDA expectations for design control. Jama Connect provided a traceability functionality that worked well for our needs. In addition, the collaboration capability of Jama aided us greatly in the definition of our requirements.
How do you measure success for this project? How are you using Jama Software to get there?
Hiltner: Now that we have that [FDA] clearance on the EnVisio Navigation System, success will mean surgeons utilizing our product to improve patient care.
Guericke: Confidence in assuring complete coverage from customer requirements to product requirements to software requirements to verification and validation. I really like how intuitive Jama Connect is. It was very easy for me to figure out how to navigate and use. There was some learning curve on the system configuration, but the support provided by the Jama Software team made that much easier.







: Four Key Development Best Practices")
