Tag Archive for: Requirements & Requirements Management Page 13
Tag Archive for: Requirements & Requirements Management
Jama Software is always looking for news that would benefit and inform our industry partners. As such, we’ve curated a series of customer and industry spotlight articles that we found insightful. In this blog post, we share an article, sourced from MedTech Dive, titled “FDA Seeks Feedback On Health Equity for Medical Devices” – originally published on August 6, 2024, and written by Nick Paul Taylor.
FDA Seeks Feedback On Health Equity for Medical Devices
The agency plans to develop a framework for when devices should be evaluated in diverse populations to support marketing authorization.
Dive Brief:
The Food and Drug Administration is seeking feedback on health equity for medical devices to inform a potential regulatory approach to the topic, the agency said Monday. The paper is open for comment until Oct. 4.
In the discussion paper, the FDA shared its thinking on how sponsors can select study populations that adequately reflect the intended use for a particular medical device.
The discussion paper is part of a broader focus on health equity, one of the Center for Devices and Radiological Health’s strategic priorities, that also includes advice on diversity action plans.
The FDA framed the discussion paper as a response to the “urgent public health need for innovative technologies that help to reduce barriers to achieving health equity.” Seeking to address that need, the CDRH has committed to developing “a framework for when a device should be evaluated in diverse populations to support marketing authorization” as part of its strategic focus on health equity.
Running clinical trials that generate results consistent with how a device will perform in the real world is a way to improve health equity. Acknowledging that generating clinical data “can be complex,” the agency said it has focused its discussion paper on “a few important considerations that may be relevant for FDA’s evaluation of clinical evidence.”
The paper outlines factors that may help sponsors and investigators develop study objectives. To inform early trial design, the FDA recommends asking how the burden of disease and how the prognosis of a disease varies across a device’s intended use population. Sponsors can also ask how a device may introduce, exacerbate or mitigate differences in outcomes across the study population.
Another section of the document describes considerations related to the FDA’s evaluation of safety and effectiveness. The FDA reviews marketing authorization filings to determine whether there is reasonable assurance the device will be safe and effective in the intended population. As such, the agency is looking at whether clinical data “are generalizable to, and representative of, the intended use population.”
The FDA said it “considers it important to understand a sponsor’s rationale regarding the relevance of the provided clinical data to the intended use population.” The rationale could cover the processes used to select and enroll study populations, the agency said, or help the FDA understand difficulties a sponsor encountered when trying to obtain data from certain populations.
In the final section of the paper, the FDA describes example scenarios intended to prompt feedback on its assessment of the benefits and risks of devices. The section features a table that shows how the FDA may respond depending on whether the evidence suggests there are differences in patient populations and health outcomes, as well as whether the sponsor included specific populations in the clinical study.
In some cases, clinical trial sponsors may choose to design studies with “enriched” populations, meaning they intentionally enroll people from groups where differences are expected. The table suggests sponsors that fail to enrich study populations to reflect the intended use may face difficulties at the FDA if the available information suggests differences in patient populations.
The FDA said the absence of enriched populations may “introduce uncertainty” on the applicability of the data to the intended use population.
In this blog, we recap our webinar, “Application of Systems Engineering in Healthcare”. Click HERE to watch the entire webinar.
When it comes to healthcare, the first to market usually gains 80% of the market share, making development speed one of the most crucial aspects of success – or failure. That’s why many organizations are looking at systems engineering as a way of connecting needs to solutions.
In this webinar, Chris Unger of PracticalSE LLC and Vincent Balgos Director, Medical Device Solutions at Jama Software® have partnered up for an engaging webinar on the application of systems engineering in healthcare.
We invite you to join in as we delve into transformative role systems engineering is playing in the healthcare industry.
What to Expect:
1. The Power of Simplicity:
Discover how focusing on the basics, while maintaining world-class performance levels, can yield astonishing returns. We’ll show you how simplicity can be a game-changer in the complex world of healthcare systems engineering.
2. Market-Driven vs. Contract-Driven:
Intrigued by the difference between market-driven and contract-driven industries? We’ll explore how systems engineering varies in these two landscapes. Learn why “Market Driven” industries emphasize competitive value creation and use cases more than traditional requirements, and how this shift can redefine your approach in healthcare.
3. Striking the Perfect Balance:
Explore the ideal state of systems engineering in healthcare, often a harmonious blend of Agile, Lean Startup, and more traditional systems approaches. Uncover strategies to adapt, innovate, and succeed in this dynamic field.
Don’t miss this opportunity to gain a comprehensive understanding of how systems engineering can revolutionize healthcare. Whether you’re a seasoned professional or just beginning your journey in healthcare systems, this webinar promises valuable takeaways for all.
Below is an abbreviated transcript of our webinar.
Application of Systems Engineering in Healthcare
Chris Unger: We’re going to talk today about systems engineering in the medical industry, particularly medical device development. So the medical device industry faces several challenges. There’s clearly constant time pressure in developing and launching safe and effective products. We’ve got to be faster than the competition with better products. And as you can see from the statistics, this is a challenge. Part of the challenges in delivering things on time is the shifting regulatory landscape. I’m sure everyone’s aware of MDR. There’s software for medical devices. The FDA is going to think about redoing design controls next year. When we were at GE Healthcare, there were like 8,000 regulations we had to monitor. So it’s a very challenging and shifting regulatory landscape. Not only do you have to be compliant with regulations, but you have to ensure your device is safe. And so quality issues, safety and just keeping performance are key elements of delivering on time and that’s getting more and more expensive as you can see here, billions of dollars of financial risk of getting this wrong.
So to make all that harder, there’s a constant increase in complexity. When I first started, there were typical software development teams were 20 to 40 people. It’s now hundreds of people and lots of interactions. So additional things like AI, machine learning, or new technologies, really have to manage a lot of complexity inside of your devices. The organizational structure is getting more and more complex. There’s a heavy focus on acquisition, so you’re integrating new teams, new cultures, and geographically distributed development teams. So that makes it all challenging. So we’re going to talk about how systems engineering can help address some of these particular challenges.
Unger: As I mentioned, a key differentiator is getting to market faster. So the success of a program in a market-driven environment is basically profits. The first mover tends to collect the lion’s share of the profits. We typically have many customers. You don’t have a single customer marketing and product management tells you roughly what they think the competition will be and what differentiates versus in a contract-driven environment, success is satisfying the contract. So within GE Healthcare, the avionics and oil and gas businesses typically had a single customer. We would produce a floating city block to British Petroleum or Shell, go to the North Sea or the Caribbean and you had a contract and you delivered to that contract versus an engine, an aircraft engine, or a medical device, we deliver to the marketplace. We decide the timing, we decide the features.
So the stakeholders and market-driven are internal to the business and you can negotiate budget and time. If you get a really, really cool feature, you can take an extra month or quarter to develop it, versus in a contract-driven, it’s really fixed. So the challenges of market-driven and contract-driven are different. Contract-driven requirements are a key commitment. You’ve got to negotiate a formal design control versus within a market-driven environment they’re critical. You have to deliver validated requirements, but they’re definitely an internal business tool that helps communication across all the business functions.
So what’s the value of systems engineering in a market-driven industry? We basically turn the ambiguous needs that we get from product marketing or product management and turn them into clear and feasible solutions to be implemented by the hardware and software teams. The key value we produce is that those implementations seamlessly integrate into the customer’s workflow and work systems. So they work really well from day one, they reliably meet their needs. They work really well after five years and not just meet their needs, they delight the customer. We really want to deliver something that the customer enjoys using. So we have to make it work day one, we need to make it work day 50. We need to make it work for every single customer. So you have to deal with all the known variability of hardware and process. Every installation and every service event has to produce a uniform, high-quality, high-performing product.
So with those constraints, we want to optimize the business value. So when we have multiple options, marketing will tell us the customer value of these options. The implementation teams will tell us the delivery and product cost of those functions. The role of systems engineering is to make trade-offs between those and really optimize the business impact based on the cost of implementation. So we want to make sure the work done by those implementation teams is tied to the maximum market impact. And associated with that is managing technical risks. If you go down a path and it turns out to be infeasible, while it might’ve been nice if it worked, you just wasted a lot of that work. So that cost has to be scaled by risk.
In doing all those first four bullets, our key value is making sure design decisions are identified and closed predictably, and that the team acts with one voice. So decisions are framed, the options are agreed to, the decision criteria are agreed to and the final decision is closed and stays closed as stakeholders change. So once you have a frozen design, do you want to make sure that actually integrates easily and when you have integration or quality problems, they’re found early and resolved early. When you have time to react, it’s a lot easier to adjust your design in the first half of your program. It’s really hard when you find severe quality issues with a month left before shipping.
Unger: And so really winning products happen when systems thinkers are effective. So clearly there’s going to be a need for some systems engineering process thinkers, but they’re system thinkers across the entire program. And so we want to make sure that everybody’s involved in systems and that the creativity of the entire program is maximized. So getting specific to GE Healthcare, what is systems engineering at GE Healthcare? Well, we have the essential customer-perceived performance. So a lot of our programs are imaging, so we have the image quality. Still, we also have things like maternal-infant care where we deliver the right humidity and temperature around the neonate. In delivering that essential performance, you’ve got to make sure it’s safe and you’ve got to make sure you have regulatory compliance. And I mentioned we really want products that are easy to use and delight the customer. So usability is a critical part of systems engineering. In doing that, we make sure we define the right implementation requirements and the right reliability strategy, and that it can be installed and serviced properly.
So with that being the overall goal, how do we organize? Well, there are a lot of things that are common across all of our product teams. We do have common program milestones. We do have a common systems’ lifecycle. It’s basically the V-cycle with iteration and agile built in. What differs is that different product teams at GE Healthcare have different levels of safety hazards, so FDA risk. We go from anesthesia where you can easily kill somebody down to ultrasound, where it’s non-ionizing equipment, that’s the light handheld probes. You can’t pinch or crush anybody to even service software that has zero patient impact. There are also almost no risks for anybody and we respond to that by adjusting the process rigor so that the higher-risk safety risk modalities have higher process rigor.
Additionally, things vary across the world or we have different locations with different cultures and different sizes of organizations. We have many systems engineers across the company, but the SE team sizes vary from less than 10. In fact, we had some sites with maybe 10 engineers and the systems engineer was half a person to teams that had over a hundred systems engineers. The scale of the programs we work on is less than 10 engineers and months-long programs to many hundred as engineers applied to a program that might last three years and were based on technology developed over the prior decade. And you want some systems engineering thinking even during that basic research decade.
Unger: The organization goes from product centralized, it’s like the SE GM for that hundred engineering group where they all reported to a dotted or solid line, to decentralized in where that team of 10 with one or half a systems engineer, there the manager was a general engineering manager and did not have a lot of systems engineering experience. So I joke that if there is a way of organizing systems engineering, we have one of those within our group somewhere.
But how did we think about tailoring? And so this is a page I put together that was generalized that you might be able to use. Obviously, as I mentioned, higher technical risks including safety risks. One way of measuring that is how many risks there are in your hazard analysis. For things that are a higher risk we looked for a higher level of functional excellence, more process documentation, more process compliance, and higher rigor of the technical design reviews, and maybe more independent reviewers. Team experience. This is subjective to measure, but Joel Goldstein did a very nice study, from Carnegie Mellon, that the value of systems engineering increases with program complexity, but it decreases with a more experienced team if you have a small team that is experiencing the technology and the application, they can get by with less process rigor and while systems engineering excellence delivers some value, it delivers less value.
Proactive and Live Traceability™ in Jama Connect® vs. Retroactive and Lagging Traceability in Excel
Incremental increases in product development complexity can lead to an exponential increase in the effort required from Engineering and Research and Development teams to keep up in a document-based (Word/Excel) environment.
PROBLEM
Our medical device & life sciences team has thousands of conversations per year with people interested in product and systems development improvements.
When we look at customer data over the two years prior to adopting Jama Connect, 83% of these organizations had their traceability maintained in an Excel-based matrix.
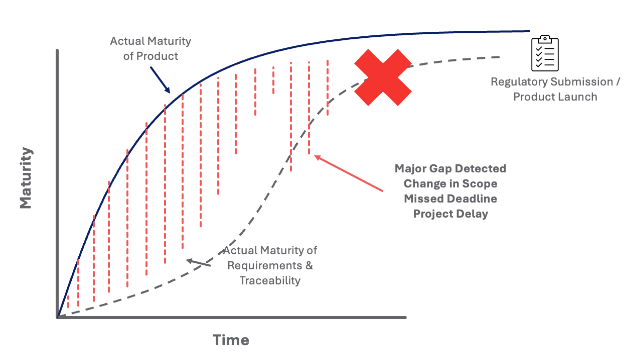
In this environment, all of the traceability components are maintained in a separate system (in other documents or tools). What we found was that this led to traceability being disconnected from the actual design. And in these cases, this delta was maintained and updated manually.
Updating the Excel-based traceability matrix to reflect changes or new artifacts is always a manual process.
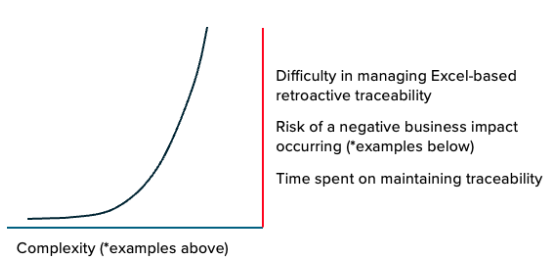
Because this process is not automated, it takes a significant effort and is also highly error-prone. On a small scale, it can be manageable. But once a change is made, managing it effectively in this way becomes a significantly greater problem. We found that these events can exponentially increase both the level of effort needed to maintain traceability and the risk of a negative outcome or occurrence. We’ll provide some examples of this shortly.
As organizations make incremental improvements and changes, or grow and scale the company further, the manual, Excel-based process can become a major bottleneck. Because the level of effort associated with changing this workflow can seem too big of a task to complete, process improvements are de-prioritized. We see an exponential scale difference between an increase in complexity and the difficulty in managing traceability in Excel manually – tightening this bottleneck. Even a slight increase in complexity can lead to a high-severity issue/business impact/time waste.
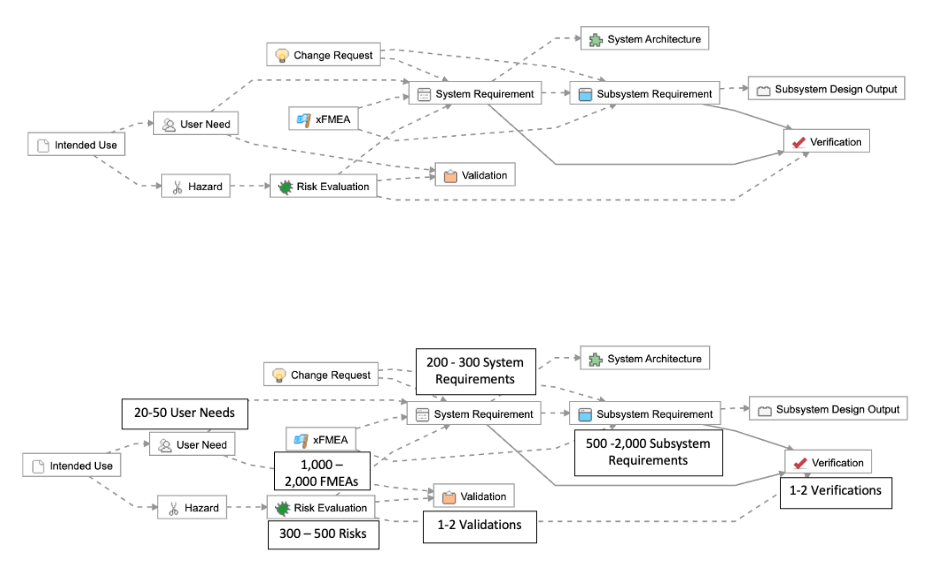
As an example, let’s take a simpler medical device (a single-use catheter). Here are a few examples of how you could have the traceability schema established:
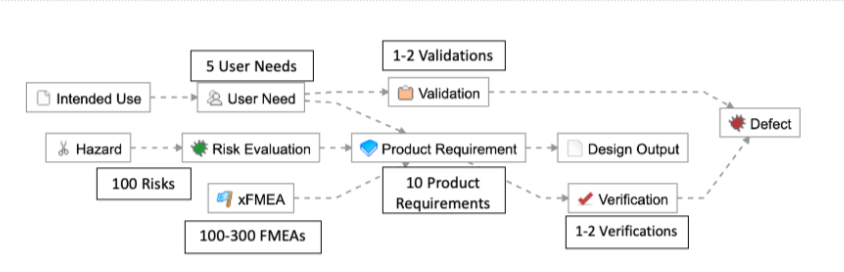
1-2 levels of requirements traceability (e.g., User Need > Product Requirement > Verification, or User Need > Validation)
Few items per level at each level in the hierarchy (e.g., 5 User Needs, 10 Product Requirements, and 1-2 Verifications/Validations)
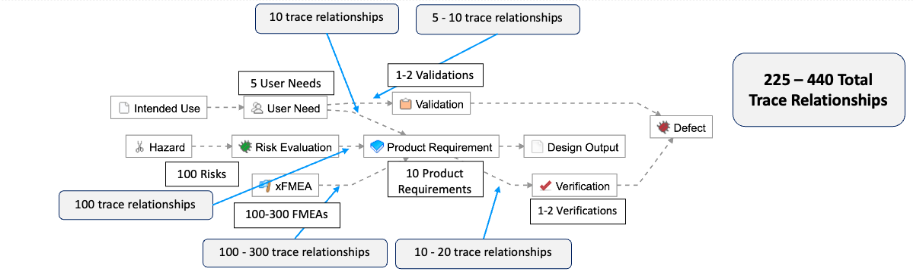
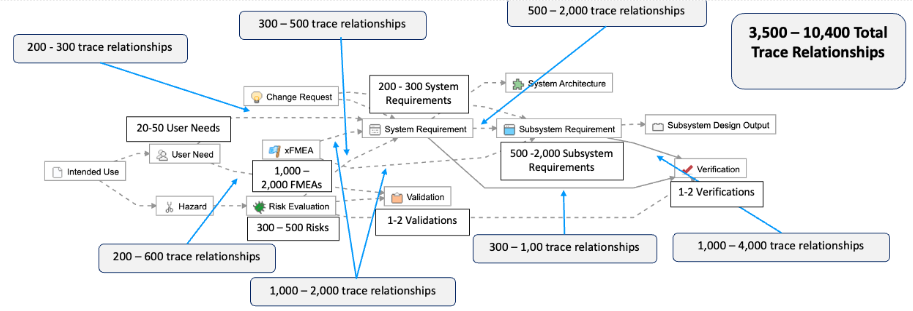
As described in the example below, this means for simple products like a single-use catheter, there are roughly 225-440 possible trace relationships:
Let’s now imagine that we want to make a seemingly simple change and additional functionality to the device. We want to connect the catheter to a mobile application, so it can monitor its usage and analyze it for diagnostics purposes.
As this device is now part of a “system” with two major sub-system components, we break down the “product requirements” into two separate levels: “system” and “sub-system” requirements.
The complexity associated with managing requirements and maintaining traceability increases exponentially. For example, we’ve outlined a common example of a Class II system, where you can see that a 4x – 10x increase in the number of User Needs translates to a 15x – 24x increase in the total number of trace relationships that need to be managed.
An increase of 4x-10x in the number of User Needs translates to a 15-24x increase in the total number of trace relationships that need to be managed.
Here are a few common real-world scenarios where we see this complexity change occurring; A couple of examples of other complexity increases for a medical device organization:
A medical device with a newly added component and functionality (software, electrical, mechanical, hardware, etc.)
A research use application (RUO) that is reclassified as an IVD – becoming a diagnostic device
A medical device startup going from an R&D phase into having a product out in the market
A medical device being implemented into a larger system
Expanding into different markets and adhering to new regulations (US FDA, EU MDR, etc.)
Introducing product families, product lines, and variations to more effectively reuse existing components
The more this process/tool improvement is delayed, the higher the risk to the business.
We see this happening to medical device companies both big and small. Here are a few of our customers describing the problem.
“With Word and Excel, if something is changed and a link is broken, that document is gone and it’s literally floating around somewhere in the cloud without linkage to anything. This makes it very scary, especially from a quality or regulatory perspective. Our Word and Excel process evolved with the organization and therefore it was put together layer by layer, making it really hard to have the full depth of knowledge about how the quality system works.” – Rene Wenmekers, Director of Quality & Regulatory, Microsure
“We work in a highly regulated environment, and Microsure’s product has hundreds of requirements on system, subsystem unit, and component levels. And from a regulatory documentation standpoint, information is scattered.” – Robin Brounds, Software Team Lead, Microsure
“When we make changes in medical device development, they need to be reported to the notified body. And when that change hits the level of ‘significant change,’ the whole documentation set needs to be provided to the notified body to be reassessed on safety and efficacy. Every time a requirement changed, it needed to be updated across the whole documentation path. This was not sustainable using Word and Excel, and it was risky.” – Rene Wenmekers, Director of Quality & Regulatory, Microsure
Convergent Dental
While using Word and Excel, Convergent Dental found themselves tracking across multiple documents, all with their own trace matrix tables relating to different requirements. The fallout from this process is that even a single word or letter change in a low-level subsystem requirement led to updating corresponding requirements documents in their trace matrix tables. So, a single letter turns into not one change but potentially six changes across five different documents.
“We have a small team with a large amount of features and updates to perform on an ongoing basis. We all work really hard here, and there’s no option to be dead weight. Getting rid of that wasted time in Word and Excel and getting our test engineers back to work is the ultimate goal.” – Craig Woodmansee, Electrical Systems Engineer, Convergent Dental
NEGATIVE BUSINESS IMPACTS – of not changing
Wasted Time and Inefficient Processes
In a complex setting (working with a complex product, highly regulated environment, high-risk product, cross-functional teams working together, lots of different product variations, target customers/markets, etc.), this can be a person or a team’s full-time effort to keep up to date.
Time is being spent on people trying to find the right and most up-to-date documents
Sitting through review and alignment meetings with all stakeholders
Increased Risk of Negative Outcomes
This process relies on people constantly monitoring and updating each change. If a change goes unnoticed or people forget to update traceability, this gap is difficult to notice. If traceability gaps are noticed later during the product development lifecycle, there is a significant increase in the risk of one of the following negative events happening:
Releasing a faulty & untested product, quality compromise, product callbacks
Forcing organizations into late-stage changes that are costly to implement
Regulatory issues and audit findings (non-conformities, FDA warning letters, etc.)
Product not meeting the original requirements and customer/stakeholder needs
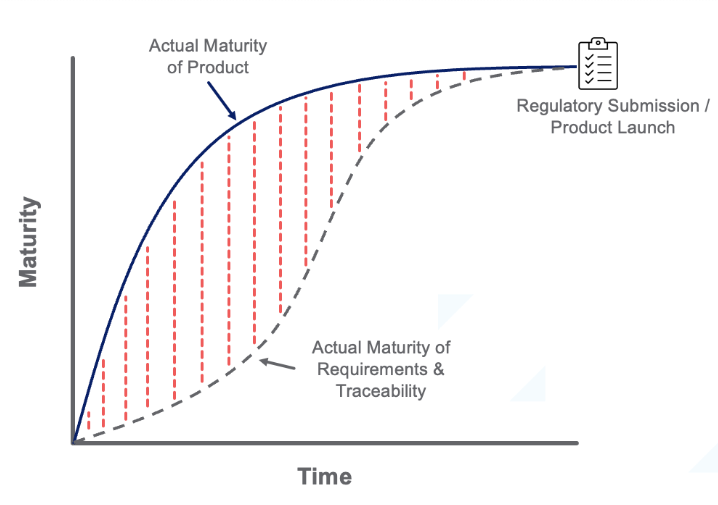
The expected outcome – backfilling documentation and traceability at the very end of the project.
Real outcome – Many issues/gaps went under the radar, leading to project delays, missed deadlines, or regulatory/quality issues:
Decreased Organization Maturity, Disconnected and Siloed Teams
Enforce the defined process – In a document-based environment, it’s close to impossible to monitor and enforce the defined process
Impact on employee tenure – Engineering and R&D are forced into manual documentation instead of actual design & development
Impact on talent acquisition – High-quality talent is more attracted to companies with proper tooling and processes in place
Communication and Transparency – Audit trails and change logs are often lost, hard to keep people accountable for changes
The solution to this problem is having integrated risk management with Live Traceability™ in Jama Connect®. Jama Connect will be the overarching system across all product development initiatives, bringing together all disciplines, making it significantly easier to visualize complex traceability hierarchies, replacing the manual effort needed to keep the documentation up to date, etc.
Jama Connect® brings comprehensive and detailed insights into your complex product, systems, and software development processes – automating the measurement of requirements traceability and coverage across disciplines and your organization’s toolchain.
This level of visibility helps eliminate rework due to out-of-date information; and the biggest fear for engineering leadership – that the greatest risks to a project are unseen until it is too late.
Understanding UN155 and Its Impact on Cybersecurity Management
In the ever-evolving landscape of cybersecurity, staying ahead of emerging threats and regulations is crucial for organizations worldwide. One such regulatory framework making waves in the cybersecurity community is UN155. This post aims to shed light on UN155 and its significance in cybersecurity management.
What is UN155?
UN155 is a regulatory framework established by the United Nations to enhance cybersecurity practices across various sectors. The framework sets forth comprehensive guidelines and standards for organizations to protect their information systems, data, and infrastructure from cyber threats. It emphasizes a proactive approach to cybersecurity, encouraging organizations to implement robust security measures and continuously monitor and adapt to the evolving threat landscape.
UN155 encompasses several critical components designed to strengthen cybersecurity management:
Risk Assessment and Management: Organizations are required to conduct regular risk assessments to identify potential vulnerabilities and threats. This involves evaluating the likelihood and impact of various cyber risks and implementing appropriate mitigation strategies.
Incident Response and Reporting: UN155 mandates the establishment of incident response plans to swiftly address and mitigate cybersecurity incidents. Organizations must also report significant incidents to relevant authorities, ensuring transparency and accountability.
Data Protection and Privacy: Protecting sensitive data is a cornerstone of UN155. Organizations must implement stringent data protection measures, including encryption, access controls, and data minimization, to safeguard personal and sensitive information.
Continuous Monitoring and Improvement: UN155 emphasizes the importance of continuous monitoring and improvement of cybersecurity practices. Organizations are encouraged to regularly review and update their security measures in response to new threats and vulnerabilities.
Training and Awareness: Educating employees about cybersecurity risks and best practices is crucial. UN155 requires organizations to conduct regular training and awareness programs to ensure that staff members are equipped to recognize and respond to cyber threats.
The implementation of UN155 has significant implications for cybersecurity management:
Enhanced Security Posture: By adhering to the guidelines set forth by UN155, organizations can significantly enhance their security posture. Proactive risk assessments, robust incident response plans, and continuous monitoring contribute to a more resilient cybersecurity framework.
Regulatory Compliance: Compliance with UN155 is not just a best practice; it is often a legal requirement. Organizations that fail to comply with the framework may face legal penalties, reputational damage, and financial losses.
Improved Incident Response: With established incident response plans, organizations can respond more effectively to cybersecurity incidents. This minimizes the impact of breaches and ensures a quicker recovery, reducing downtime and financial losses.
Increased Stakeholder Confidence: Demonstrating compliance with UN155 can enhance stakeholder confidence. Clients, partners, and investors are more likely to trust organizations that prioritize cybersecurity and adhere to recognized standards.
Global Harmonization: UN155 promotes a standardized approach to cybersecurity, fostering global harmonization of security practices. This is particularly important for multinational organizations operating in diverse regulatory environments.
UN155 represents a significant step forward in the global effort to enhance cybersecurity management. By adopting the framework’s guidelines and principles, organizations can bolster their defenses against cyber threats, ensure regulatory compliance, and build trust with stakeholders. As the cybersecurity landscape continues to evolve, frameworks like UN155 play a pivotal role in shaping a secure and resilient digital future.
Note: This article was drafted with the aid of AI. Additional content, edits for accuracy, and industry expertise by McKenzie Jonsson and Matt Mickle.
Jama Software is always looking for news that would benefit and inform our industry partners. As such, we’ve curated a series of customer and industry spotlight articles that we found insightful. In this blog post, we share an article, sourced from Med Device Online, titled “What You Should Know About The FDA’s New Final Rule On LDTs” – originally published on May 14, 2024, and written by Mahnu V. Davar, Philip R. Desjardins, Abeba Habtemariam, and Phillip V. DeFedele, Arnold & Porter.
What You Should Know About The FDA’s New Final Rule On LDTs
On May 6, 2024, the U.S. Food and Drug Administration (FDA or the agency) published its highly anticipated final rule, which was formally published in the Federal Register on May 6, revising the regulatory definition of an in vitro diagnostic (IVD) product to explicitly capture IVDs manufactured by laboratories.
This follows the absence of proposed congressional action and the FDA’s review and consideration of comments to the October 2023 proposed rule (described in our prior Advisory on this subject) resulting in modifications to its phaseout policy and continued exercise of enforcement discretion for certain tests. In connection with the final rule, the FDA also issued two draft enforcement policies for certain tests offered in response to emergent situations or public health emergencies (PHEs). Despite the potential for legal challenges to the final rule, clinical laboratories should begin thinking about strategies for evaluating whether their laboratory-developed tests (LDTs) are subject to the FDA’s phaseout policy, determining the extent of FDA requirements that apply to such LDTs, and engaging with the FDA.
What Did the Final Rule Do?
The final rule amended the definition of an IVD in 21 C.F.R. § 809.3 to make clear that these products are devices as defined under the Federal Food, Drug and Cosmetic Act (FDCA), and may also be biological products under the U.S. Public Health Service Act, “including when the manufacturer of these products is a laboratory.” Although not a substantial rewrite of the IVD definition, this added phrase makes FDA’s position clear that LDTs are subject to regulation as, at a minimum, devices.
Why Does This Matter?
Historically, the FDA exercised enforcement discretion for LDTs, declining to impose its device authorities over such tests in most instances. For purposes of this enforcement discretion policy, the agency defined LDTs as IVDs intended for clinical use that were designed, manufactured, and used within a single clinical laboratory certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA) that meets CLIA regulatory requirements to perform high complexity testing. As such, LDT manufacturers that generally operated outside FDA oversight will now be expected to come into compliance with FDA requirements and controls applicable to their tests. In consideration of this substantial operational and compliance burden, the preamble to the final rule details a phaseout policy under which FDA will gradually end its general LDT enforcement discretion policy in five phases over a four-year period.
The phaseout policy generally applies to LDTs as defined above, as well as “IVDs offered as LDTs,” meaning tests that are manufactured and offered as LDTs by CLIA-certified laboratories that meet CLIA requirements to perform high complexity testing and that are used within such laboratories, even if they do not fall within FDA’s historical understanding and definition of an LDT because they are not designed, manufactured, and used within a single laboratory. Thus, the policy technically applies to a broader range of tests than those that actually meet the FDA’s definition of an LDT. However, despite this breadth, the final rule makes clear that the phaseout policy does not extend to IVDs manufactured or used outside of a laboratory, including collection devices.
Consistent with the proposed rule, FDA made clear that certain tests would be excluded from the phaseout policy and subject to immediate regulation since they were never eligible for enforcement discretion:
Direct-to-consumer tests
Tests intended as blood donor screening or human cells, tissues, and cellular and tissue-based products donor screening tests required for infectious disease testing under 21 C.F.R. § 610.40 and 21 C.F.R. § 1271.80(c), respectively, or required for determination of blood group and Rh factors under 21 C.F.R. § 640.5
Tests intended for actual or potential emergencies or material threats declared under Section 564 of the FDCA
Further, the final rule confirmed that the FDA would continue exercising enforcement discretion (and thus not apply device requirements) to the following tests originally described in the proposed rule:
“1976-Type LDTs” (i.e., tests that use manual techniques without automation performed by laboratory personnel with specialized expertise, use components legally marketed for clinical use, and are designed, manufactured, and used within a single CLIA-certified laboratory meeting CLIA requirements for high complexity testing)
Human Leukocyte Antigen (HLA) tests that are designed, manufactured, and used within a single CLIA-certified laboratory meeting CLIA requirements for high complexity histocompatibility testing and used for HLA allele typing in connection with organ, stem cell, and tissue transplantation, HLA antibody screening and monitoring, or conducting real and “virtual” HLA crossmatch tests
Tests intended solely for forensic or law enforcement purposes
Tests used solely for public health surveillance when intended solely for use on systematically collected samples for analysis and interpretation of health data for disease prevention and control and the test results are not reported to patients or their healthcare providers
Notably, in consideration of comments and other feedback, FDA decided to continue to exercise full or partial enforcement discretion for the following tests:
LDTs manufactured and performed within the Veterans Health Administration or the Department of Defense (full enforcement discretion continues)
LDTs approved by the New York State Clinical Laboratory Evaluation Program (enforcement discretion continues for premarket review requirements)
Non-molecular antisera LDTs for rare red blood cell antigens where such tests are manufactured and performed in blood establishments, including transfusion services and immunohematology laboratories and where there is no alternative available to meet the patient’s need for a compatible blood transfusion (enforcement discretion continues for premarket review requirements and all Quality System (QS) requirements other than the recordkeeping requirements at 21 CFR 820 (Subpart M))
Currently marketed IVDs offered as LDTs that were first marketed prior to the date of issuance of the Final Rule (May 6, 2024) and that are not modified or are modified in certain limited ways (enforcement discretion continues for premarket review requirements and all QS requirements other than the recordkeeping requirements at 21 CFR 820 (Subpart M))
LDTs manufactured and performed by a laboratory integrated within a healthcare system to meet an unmet need of patients receiving care within the same healthcare system (enforcement discretion continues for premarket review requirements and all QS requirements other than the recordkeeping requirements at 21 CFR 820 (Subpart M))
For those tests subject to only partial enforcement discretion, the final rule makes clear that all other requirements would apply as they are phased-in under the general phaseout policy for all IVDs offered as LDTs.
What Is The Phaseout Policy?
The phaseout policy consists of the following five stages, which start on May 6, 2024:
Stage 1: FDA will end the general enforcement discretion policy as to medical device safety reporting, correction, and removal requirements, and QS complaint file requirements one year after the publication of the final rule. Thus, manufacturers of all IVDs offered as LDTs that are not subject to the FDA’s continued exercise of full enforcement discretion must come into compliance with these requirements by May 6, 2025.
Stage 2: FDA will end the general enforcement discretion policy as to all other medical device requirements, except for QS and premarket review requirements, two years after publication of the final rule. Therefore, manufacturers of all IVDs offered as LDTs that are not subject to the FDA’s continued exercise of full enforcement discretion must come into compliance with these requirements by May 6, 2026.
Stage 3: FDA will end the general enforcement discretion policy as to the following QS requirements three years after the publication of the final rule: (1) design controls (21 C.F.R. § 820.30); (2) purchasing controls (21 C.F.R. § 820.50); (3) acceptance activities (21 C.F.R. §§ 820.80 and 820.86); (4) corrective and preventative actions (21 C.F.R. § 820.100); and (5) records requirements (21 C.F.R. Part 820, Subpart M). As such, manufacturers of all IVDs offered as LDTs that are not subject to the FDA’s continued exercise of full enforcement discretion or partial enforcement discretion as to certain QS requirements must come into compliance with these requirements by May 6, 2027.
Stage 4: The FDA will end the general enforcement discretion policy as to premarket review requirements for high-risk IVDs (i.e., Class III IVDs or IVDs subject to Biologics License Application requirements) three and a half years after publication of the final rule. Manufacturers of all IVDs offered as LDTs that are not subject to the FDA’s continued exercise of full enforcement discretion or partial enforcement discretion as to premarket requirements must come into compliance with these requirements by November 6, 2027. Notably, on its media call regarding the final rule, the FDA stated that it intends to complete the reclassification of certain Class III IVDs to Class II IVDs in advance of this deadline, thus lessening the number of PMAs that will be submitted. We further discuss this initiative and its potential relation to the final rule in our related Advisory on this topic.
Stage 5: The FDA will end the general enforcement discretion policy as to premarket review requirements for all remaining IVDs requiring FDA review unless otherwise noted by the FDA (i.e., some Class I and all Class II IVDs) four years after publication of the final rule. Manufacturers of all IVDs offered as LDTs that are not subject to the FDA’s continued exercise of full enforcement discretion or partial enforcement discretion as to premarket requirements must come into compliance with these requirements by May 6, 2028.
Overview Of Test Types And Status
What About The Draft Public Health Emergency Guidance?
Although the draft PHE policies both address certain tests offered in response to emergent situations or public health emergencies, one relates to tests offered prior to issuance of a declaration under Section 564 of the FDCA while the other relates to tests offered during a declared emergency. These are notable because the FDA explains in the final rule that its general enforcement discretion policy for LDTs never applied to tests intended for emergencies, potential emergencies, or material threats declared under Section 564 because false results can have serious implications for disease progression, public health decision-making, and patient care. Instead, after all previous declarations under Section 564, the FDA has generally expected LDTs to comply with applicable requirements of the FDCA and FDA regulations. However, FDA has adopted, and may continue to adopt, specific enforcement discretion policies for such tests.
FDA’s draft Enforcement Policy for Certain In Vitro Diagnostic Devices for Immediate Public Health Response in the Absence of a Declaration under Section 564 sets forth FDA’s enforcement policy for “immediate response” tests for use in an emergent situation (i.e., the period of time between detection of the exposure or outbreak and either resolution of the exposure or outbreak or issuance of an applicable Section 564 declaration). Notably, the policy only applies to premarket review requirements and does not extend to tests with home specimen collection or at-home tests.
Under the policy, FDA does not intend to object to the offering of “immediate response” tests when:
The test is manufactured and offered by laboratory manufacturers meeting certain criteria
The test has been appropriately validated
FDA is notified
Appropriate transparency is provided
The test is labeled for prescription use only
There is no applicable Section 564 declaration
If no applicable Section 564 declaration is made within 12 months of the start of an emergent situation, FDA anticipates that the public health rationale for the enforcement policy will no longer apply at that time. FDA would then expect the laboratory manufacturer to cease offering the immediate response test or seek approval/clearance/authorization. FDA does not intend to object to the continued offering of an immediate response test while the premarket submission is prepared, submitted, and under FDA review so long as the laboratory manufacturer submits the submission within 12 months from the date of the first offering of the immediate response test.
FDA’s draft Consideration of Enforcement Policies for Tests During a Section 564 Declared Emergency, when finalized, will describe the factors FDA plans to assess in deciding whether to issue an enforcement policy regarding test manufacturers’ offering of certain devices (i.e., unapproved tests and unapproved uses of approved tests) for the diagnosis of disease or other conditions during a declared emergency. FDA intends to assess, among other things:
The need for accelerated availability of such tests
The known or potential risks of such tests
The availability of appropriate alternative tests that are authorized or approved
The availability of sufficient mitigations to address the risks of false results
FDA intends to conduct webinars, publish guidance documents, provide templates, and participate in conferences to help laboratories understand and comply with applicable devices. We also think it likely that the FDA will increase inspections of laboratories offering IVDs as LDTs where the agency has identified or received concerns regarding their quality or accuracy and will start enforcing the FDCA against laboratories and similar entities perceived to be abusing the agency’s Research Use Only/Investigational Use Only policy or not complying with Investigational Device Exemption (IDE) requirements. As we noted in our Advisory on the October proposed rule, the FDA also has identified a number of test types that the agency believes present a higher risk of patient harm when run as LDTs (according to the agency, these include tests such as non-invasive prenatal screening).
Pre-submission meetings, pre-IDE meetings, and other FDA engagements related to data generation, regulatory pathway clarification, and classification will likely increase. The industry will need to closely watch the steps the FDA takes to lessen the burden of the final rule, such as the noted reclassification process, as well as any potential personnel or structural changes, and future funding requests. As the final rule preamble discussion notes, the alignment of the phaseout policy to coincide with the next round of Medical Device User Fee Amendments reauthorization suggests that the agency understands that it will have to carefully evaluate the burden of this exceptional expansion of FDA authority in terms of protecting the public health while not slowing down the availability of key diagnostic advancements to aid patient care.
We anticipate that laboratories and other affected entities will consider pursuing legal action against the agency, arguing that the FDA lacks authority to regulate LDTs and seeking to enjoin the agency from implementing the final rule. The preamble discussion attempts to anticipate and resolve a number of the key challenges raised in comments to the proposed rule, such as important legal questions about FDA authority under the Federal Food, Drug, and Cosmetic Act, interstate commerce concerns, limits on regulation of state-licensed actors, and many other salient issues. FDA clearly is of the view that public health exigencies outweigh the litigation risks, and the final rule phaseout policy and other enforcement discretion positions are sufficient to balance the interests of industry, patients, and the agency.
It remains to be seen what actions Congress may take now that the FDA has articulated a final position on this topic. Although Congress has taken an interest in the regulation, and lack thereof, of LDTs, including holding a recent hearing, a legislative solution that would potentially supersede the final rule is uncertain, if not unlikely in the near term. Therefore, given the current state of affairs, it is important for laboratories offering LDTs to begin strategizing on how they will address the final rule and FDA’s phaseout policy.
Jama Software is always looking for news that would benefit and inform our industry partners. As such, we’ve curated a series of customer and industry spotlight articles that we found insightful. In this blog post, we share an article, sourced from Engineering News-Record, titled “ENR 2024 Top 500 Sourcebook: Plant Tests Ocean Carbon Dioxide Removal” – originally published on July 16, 2024, and written by David Godkin.
Arup begins feasibility study of commercial-scale facility set for Canada site
Engineering has begun on a project its developers say could mark a new foray into ocean-based carbon dioxide removal.
The Quebec-based plant would be North America’s first commercial-scale, ocean-based carbon dioxide-removal facility, according to startup developers Equatic Inc., based in Los Angeles, and Deep Sky Inc., a Montreal firm. They say the facility would remove nearly 110,000 tonnes of carbon dioxide from the atmosphere annually—10% of which will be ocean-borne emissions. It also would produce 3,600 tonnes of hydrogen.
The plant technology uses a seawater electrolysis process developed at the UCLA Samueli School of Engineering’s Institute for Carbon Management. The system draws ocean seawater into an electrolysis chamber where carbon dioxide molecules are separated from the water and its natural acidity is neutralized. “This in turn allows the ocean water to draw down more CO2,” says Phil De Luna, Deep Sky chief carbon scientist and head of engineering. “These carbon emissions are some of the hardest to abate and most difficult to electrify.”
Extracted carbon dioxide is stored in the form of solid calcium and magnesium-based substances that the firm says could be used to produce construction materials.
The technology is based on a $20-million demonstration plant, called Equatic-1, which is set to commission next year in Singapore in collaboration with its national water agency, the developers said.The commercial plant will have easy access to the ocean-connected St. Lawrence River, and could tap into Quebec’s massive hydroelectric grid.
The facility would use less than 1.4 MW hours per ton of carbon dioxide, with removal cost estimated at less than $100 per ton by 2030, according to Equatic. Luna estimates the project’s cost at about $366 million. The plant may also be in line for carbon removal credits under a new U.S. Energy Dept. program, Equatic says, noting that such credits, as well as green hydrogen from this plant and future ones “have been pre-sold to companies such as Boeing, and further sales are ongoing.”
Engineering firm Arup has just begun a six-month plant feasibility study that is set to launch front-end engineering design work, environmental assessment and permitting, with a final investment decision estimated at the end of 2026.
If affirmed, construction on a 30-acre site would begin shortly after, with startup set for 2028. Deep Sky is currently exploring a plant location about 900 km northeast of Montreal, with a final site decision to be made by the end of 2024, the firm says.
Jama Connect® Features in Five: Cameo Systems Modeler Integration
Learn how you can supercharge your systems development process! In this blog series, we’re pulling back the curtains to give you a look at a few of the powerful features in Jama Connect®… in about five minutes.
In this Features in Five Integration Series video, Gary Hayes, Senior Solutions Architect at Jama Software® – will demonstrate the Cameo Systems Modeler integration with Jama Connect® using Intercax Syndeia.
VIDEO TRANSCRIPT
Gary Hayes: Hello, and welcome to the Features in Five Integration series. My name is Gary Hayes, and I am a Senior Solutions Architect at Jama Software. Today, we will be walking through the Cameo Systems Modeler integration for Jama Connect. We make it possible for you to integrate Jama Connect with preferred best-of-breed software to achieve Live Traceability™ across the end-to-end development cycle. Live requirements traceability is the ability for any engineer at any time to see the most up-to-date and complete upstream and downstream information for any requirement, no matter the stage of systems development or how many siloed tools and teams it spans. This enables significant productivity and quality improvements, dramatically reduces the risk of product delays, cost overruns, defects, rework, and recalls, and ultimately results in faster time to market.
Let’s start off today by looking at the two environments that we’ll be working with, Jama Connect and Cameo Systems Modeler. In Jama Connect, in our project here, we have a folder called swarming along with five requirements we’ve identified that we wanna be using. If we look over the Cameo System Modeler, you notice that we have the same folder, but we only have four requirements listed there.
Hayes: We can easily compare the items that exist in both environments, the four that you see here. We go into the plug-in that we’re using, identify the SysML repository, and we drill down on that folder that contains those requirements, and we can do a comparison between the source and the target. The plug-in will do the comparison for us. We don’t have to drill down and do a close examination.
When we get our results, we see that everything is green, indicating to us that the items in the folders that have been synchronized indeed match at this point. We can close this out for the time being, but you’ll keep in mind that we only have four requirements in Cameo, but we have a fifth one that does not exist currently in Cameo that is in Jama Connect. So we want to make sure that both environments do indeed match, and we can do that easily by dragging and dropping using that same plug-in.
We go back into our dashboard. We find our SysML repository, and we find our Jama Connect project that we’re working with. Drill down on that to find those requirements that currently are being synchronized between the two environments. We can easily see that we have the swarming folder along with its five requirements from Jama Connect and four from our Cameo environment. And to match those up, we want to drag and drop this into our Cameo environment.
And you’ll notice over here as it brings that over, you notice in the background, the cameo environment updates automatically to reflect the fact that we’ve brought a new requirement into the cameo environment. We can further confirm that by doing a synchronization check, doing that comparison once again at the folder level, compare our source and target, and hopefully, we’ll get all green one more time to show that the environments do indeed match up. But we don’t always have the luxury of dragging and dropping and never making any changes in any environment, so what we’ll want to do is make a change in one environment and push that from one side to the other. So let’s go into this individual requirement, broadcast to Swarm, and it’s annotated that it is indeed from Jama Connect. So we’re gonna remove that annotation in its title.
Hayes: We’ll go ahead and save that in our Jama Connect environment. You’ll notice that’s updated. Now we want to be able to show that same type of update. We’ll do that comparison first to see where we’re at because we never know when changes will occur. We can do our compare, make sure that comparison actually works, and flag us for a change. And indeed, we do. It comes up as pink or red, depending on your monitor, and flags us that there has been a there’s a discrepancy between the two environments. And you’ll notice too that it does the comparison. It doesn’t automatically make the change, and you can see that in the background. And in our Cameo environment, that change has not rolled over from Jama Connect to Cameo. So let’s make that change, permanent now. Let’s go ahead and do that push. We can push from our target to our source.
Keep your eyes on the Cameo environment in the background. As we make that change and it gets pushed over, you’ll notice that the name or the description of the requirement in Cameo indeed has changed, and so that has been updated automatically for us. We can do one last check with our compare tool, comparing source and target. So we get all green just for one additional factor of confidence that we get there, and you can see it there. So that’s one way to keep your Cameo and Jama Connect environments in sync using a plug-in.
Thank you for watching this Features in Five session on the Cameo Systems Modeler integration for Jama Connect. If you’re an existing customer and want to learn more, please reach out to your customer success manager or consultant. If you’re not yet a client, please visit our website at jamasoftware.com to learn more about the platform and how we can help optimize your development process.
In this blog, we’ll recap our recent webinar, “Excelling in Requirements Management for Successful Software Delivery and Implementation” – Click HERE to watch it in its entirety.
Excelling in Requirements Management for Successful Software Delivery and Implementation
Are you interested in understanding the fundamentals of effective requirements gathering and analysis for the delivery of software?
In this webinar, Steven Meadows, Principal Solutions Lead at Jama Software®, discusses the challenges posed by traditional document-centric requirements processes to support customer-focused projects.
You’ll gain an understanding of:
Best Practices: Transitioning to more Agile and collaborative requirements management approaches to enhance project execution.
Case Studies: Real-world examples highlighting successful adoption of modern requirements management practices in vendor implementation projects.
Key Metrics: Learn how a requirements management platform can help improve project metrics for customer implementations.
Below is a preview of our webinar. Click HERE to watch it in its entirety.
The following is an abbreviated transcript of our webinar.
Meadows: Effective requirements management not only accelerates project timelines but also improves stakeholder collaboration and reduces the risk of project deviations.
An integral part of today’s discussion will be a deep dive into Jama Connect, a powerful tool designed to facilitate comprehensive requirements management. We’ll explore its features and functionalities that enable teams to capture, trace, and validate requirements throughout the implementation of software.
Whether you’re new to Jama Connect or seeking to optimize your current usage, this segment will provide valuable insights into leveraging the platform effectively.
Lastly, I’ll describe a case study that illustrates a successful implementation of requirements management strategies. This real-world example will demonstrate how an organization has overcome challenges, implemented best practices, and achieved tangible benefits using advanced tools and methodologies for the implementation of solutions.
By the end of this webinar, you should gain actionable insights to enhance your approach to requirements management, ultimately driving greater efficiency and success in your software delivery projects.
Now, before we get started, I’d like to briefly introduce myself and my background. With a robust background in requirements management, I bring over 10 years of experience in implementing software solutions across a broad spectrum of industries, successfully managing complex project engagement.
Throughout my career I’ve had the privilege of working closely with incredibly innovative and life-changing organizations, helping them navigate the intricate landscape of software implementation and delivery.
From defining clear and actionable requirements to optimizing workflows and ensuring seamless collaboration across teams, I’ve witnessed firsthand the transformative impact of effective requirements management.
Meadows: At Jama Software my focus has been on empowering teams to achieve their project goals efficiently and with precision. Whether it’s harnessing the full capabilities of Jama Connect or strategizing for complex project scenarios, my passion lies in delivering tangible results that drive innovation and enhance operational excellence.
Today I’m excited to share insights, strategies, and practical advice that can help you elevate your approach to requirements management. Together we’ll explore key principles, delve into best practices, and then cover strategies that can empower your organization to excel in software delivery and implementation.
I’d like to spend a moment quickly introducing Jama Software, the company that I represent. Jama Software provides the sweetest solutions that span the entire product and systems development lifecycle, from capturing and managing requirements traceability to enabling collaboration among diverse teams, Jama Software has engineered a platform that aligns with the evolving needs of today’s businesses.
You’ll see on this slide some of the verticals that we support, including regulated industries like medical devices and aerospace and defense, as well as pure software development and industrial manufacturing.
Some of the ways that we help our customers realize value is by reducing project cycle times, increasing process efficiency, and gaining visibility and control into implementation and development efforts.
Before we get to the main content of this webinar, I want to spend a moment just quickly defining what is meant by software implementation and delivery.
This is mainly related to the implementation of off-the-shelf software, as well as highly configurable applications. We work with a lot of vendors who implement their proprietary software for their customers, in particular, we have several customers in the benefits and HR space, healthcare, as well as other regulated industries including non-regulated industries. Although we’ll be touching on software development throughout this webinar, we’ll mainly be focusing on the implementation activities of applications, the challenges that come along with the implementation, and best practices to mitigate issues throughout the delivery of software for customers.
This has been an abbreviated transcript of our webinar.
Jama Software is always looking for news that would benefit and inform our industry partners. As such, we’ve curated a series of customer and industry spotlight articles that we found insightful. In this blog post, we share an article, sourced from AECMagazine, titled “Cyberattacks: safeguarding contractors” – originally published on May 22, 2024, and written by Ben Wallbank.
Cyberattacks: Safeguarding Contractors
It’s every construction firm’s biggest nightmare: criminals taking control of their data and holding them to ransom. Ben Wallbank, Trimble, shares some best practices to mitigate cyberattacks
Cybersecurity and cybercrime often conjure up images of hackers in dark hoodies, sneaking in the digital back door. In reality, nearly 90% of corporate cybercrime, such as phishing or ransomware attacks, is a result of employee error.
The UK construction industry is no exception and could be an even greater target than other industries. Protecting massive amounts of data, including warranty and latent defect remediation periods, makes contractors attractive to cyber criminals. Cybersecurity is so crucial to construction that the National Cyber Security Centre produced a construction industry-specific guide, along with the Chartered Institute of Building (CIOB).
Cybercriminals who target the construction industry usually do so by accessing, copying, and sharing data illegally or by installing malware on a company’s computers and network, taking control of files, and holding them for ransom. It’s called ransomware, and it’s probably the most common and one of the most debilitating types of cybersecurity breaches in the construction world.
Each year, we hear of new cyberattacks, taking critical infrastructure offline and crippling construction businesses worldwide, including many here in Europe. These attacks cost billions of pounds a year and can cause whole cities, businesses, and services to grind to a halt.
UK contractors should follow these best practices to safeguard against cyberattacks and improve outcomes in case of an attack.
Create a business continuity plan
Preparing for the worst puts your business in the best position moving forward because you can act quickly and have more control of the outcome. A solid cyber security disaster plan can get quite detailed. It should be consistently reviewed, practiced, and updated to net the best results in case of an incident. At a minimum, a business continuity plan should include the following:
Name of a leader to act as a central resource to manage disaster recovery across multiple departments.
A communication plan for sharing key messages and managing crises with employees, clients, and additional project stakeholders.
A maintenance plan for a continually updated (and backed up) list of employee contact information and asset inventory.
A crucial aspect of any good cyber security plan is to make sure that everything is backed up, preferably on the cloud or physically on an offsite server that’s not on your network. Backups should be frequent and automated, so ask your IT provider to set them up so that they either happen in real-time (if you’re backing up to the cloud) or that they run daily after everyone has left the office.
Secure mobile devices
Mobile devices are more challenging to secure than other data systems, but just as critical. Utilizing an enterprise management platform, such as Cisco Meraki, allows you to maintain enterprise-level control over all of your devices. These kinds of platforms ensure that individual devices are still managed centrally, and contractors can limit software installation, track devices using GPS, disable devices, and more.
Protect software and servers
When it comes to software and security risks in construction, contractors should choose platforms and software providers that take security seriously. Granular permissions, user-friendly management systems, and multi-factor authentication, for instance, are all must-haves in any construction software.
By using cloud-based, connected construction software, contractors shift the responsibility of maintaining servers, ensuring SOC 2 Type II compliance, and data backup and storage. Project and business data backups happen automatically, providing daily protection, with costs often included or rolled into users’ subscription costs. New software features and security functionality are also rolled out automatically.
By coupling the backups with cybersecurity protections, cloud vendors use the latest technologies to thwart cybercriminals and provide an extra level of protection not otherwise achieved through in-house backups. When shopping for business software, make security one of your first discussion points.
Additionally, your web and email servers need to be properly protected to avoid online attacks. Physical network servers need to be secured, and you need to ensure that any cloud-based solutions you’re using also implement rigorous security protocols.
Cybersecurity protection in construction requires every employee at every level to be fully engaged and actively vigilant. There are several steps to take to make that happen:
Ensure all employees receive regular cybersecurity training, especially if online workflows or procedures change.
Welcome feedback from team members and update cybersecurity policies and processes as needed.
Counsel employees on everyday things to look for before opening email, like spelling and grammar errors, verifying sender’s email address, and never opening unexpected attachments.
Take the first step: get started
The most important step is the first one. The UK government offers two certifications – Cyber Essentials and Cyber Essentials Plus – that are crash courses in the basics to keep businesses safer from cybercrime. While they don’t replace a cybersecurity risk assessment, they will show you how to do one and how to select the security measures your business needs.
Anywhere your data is stored or used is a potential entry point into your company’s digital existence. It only takes one slip to allow malicious code or ransomware in, and once it’s there, it can cause millions of pounds worth of damage.
Jama Connect® Strengthens its Lead as the #1 Requirements Management Solution in G2®’s Summer 2024 Report
We are thrilled to announce that Jama Connect® has once again been named the overall leader in the G2 Grid® Report for Requirements Management Software for Summer 2024.
G2’s rankings are based on authentic user reviews and data gathered from online sources and social networks, analyzed through their unique v3.0 algorithm. The Summer 2024 G2 Grid® Report reflects scores calculated up until June 4, 2024.
In addition to being recognized as the top requirements management software, Jama Connect® has earned several other accolades for Summer 2024:
Overall Leader
Enterprise Leader
EMEA Leader
Europe Leader
Small-Business Leader
Mid-Market Leader
Momentum Leader
Learn more about the Summer 2024 G2 Grid for top Requirements Management Software products: DOWNLOAD IT HERE
Jama Software® is honored to receive this recognition, which highlights the value we bring to our customers, especially those moving from document-based approaches to complex product, systems, and software development. We are grateful to our customers for their valuable feedback on our product, services, and support.
Customer Feedback Highlights
“Jama [Connect] is not only a ‘document-oriented’ ALM tool, it gives the organization the ability to map the project structure the product structure making it an easy entry point for R&D folks. Configured properly, it is a real technical and regulatory ‘single source of truth.” – Frederic Fiquet, Director, Systems Engineering
“Product Design teams need a requirements management tool like Jama [Connect]. Using Jama Connect allows our software development team to have a well-organized and well-written set of requirements. It allows us to more easily maintain a baseline of features in our continuously evolving software.” — Mark M., Mid-Market
Our commitment is to provide the best possible experience for our users, and being named the overall leader is a testament to their satisfaction and success with Jama Connect.

![[Webinar Recap] Application of Systems Engineering in Healthcare](https://www.jamasoftware.com/media/2023/10/Application-of-Systems-Engineering-in-Healthcare-1.png "[Webinar Recap] Application of Systems Engineering in Healthcare")

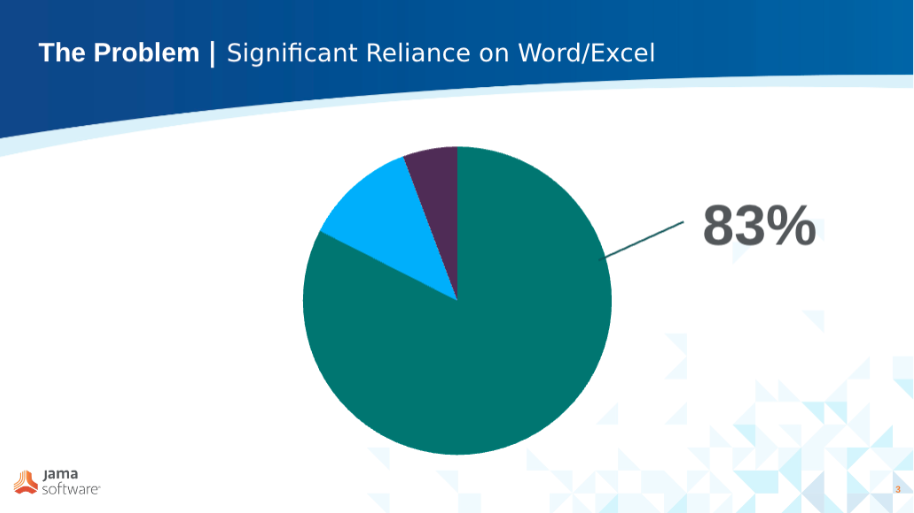 In this environment, all of the traceability components are maintained in a separate system (in other documents or tools). What we found was that this led to traceability being disconnected from the actual design. And in these cases, this delta was maintained and updated manually.
In this environment, all of the traceability components are maintained in a separate system (in other documents or tools). What we found was that this led to traceability being disconnected from the actual design. And in these cases, this delta was maintained and updated manually.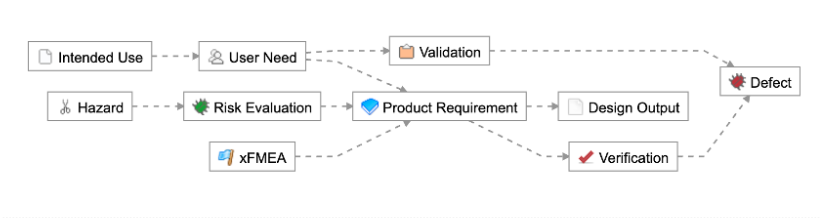






![[Webinar Recap] Excelling in Requirements Management for Successful Software Delivery and Implementation](https://www.jamasoftware.com/media/2024/07/Excelling-in-Requirements-Management-for-Successful-Software-Delivery-and-Implementation-1.png "[Webinar Recap] Excelling in Requirements Management for Successful Software Delivery and Implementation")

