Tag Archive for: medical device development Page 2
Tag Archive for: medical device development
Infographic: Jama Connect™ for Medical Device Development
We’re excited to share our latest infographic for the Jama Connect for Medical Device Development solution which explains how Jama Connect can help accelerate innovation, maintain product quality, and manage the ever-changing complex regulations in medical device development. This is a single powerful platform for medical device teams to manage design controls for device requirements and related risks, simplifying regulatory submissions, and audit preparations while accelerating time to market.
Bringing a medical device to market requires navigating a sea of complex and ever-changing regulations, not to mention bearing significant costs along the way. A device recall can cost $600 million, while the indirect costs of lost revenue and diminished market cap are even higher at $1-3 billion per company. Those costs are especially significant considering the price tag of product development—$75 million in FDA compliance alone, and an average timeline of three to seven years.
→Jama Connect customers have been able to reduce planning time as much as 80%, thanks to consolidated feedback replacing emails and a document-based approach to project management.
→Better quality products get out the door faster. By understanding the impact of change, capturing decisions and feedback in real-time and reusing existing IP, Jama Connect reduces medical device development time by an average of 130 days per project.
→Jama Software reduces rework, which accounts for approximately 30-50% of a given project and arises from issues such as requirements errors. Improving the ability to track requirements from design through verification and validation ensures teams build the right medical devices with the lowest possible lifecycle costs.
In this infographic,we share how, with the right requirements management solution, you can accelerate the development of cost-effective products that also comply with both safety and quality standards.
You’ll learn:
How to overcome the biggest challenges in medical device development
The ways Jama Connect for Medical Device Development can help
Keys to unlocking a better customer experience
In the previous blog of this series, we talked about the application of systems engineering principles to the design inputs process. In this post, we explore how the Jama Connect™ for Medical Device Development procedure guide describes connecting design inputs with subsequent processes: Design Outputs and Verifications. By supporting these processes in a single system and ensuring traceability we can build confidence in the conformance to design inputs and the proper definition of verification activities.
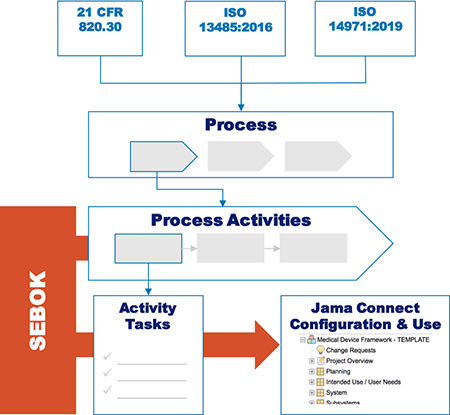
The Jama Connect for Medical Device Development solution focuses on two key aspects from the Design Output requirements defined by 21 CFR 820.30 and ISO 13485:2016 section 7.3.4. Design outputs:
must reference acceptance criteria, ensuring they are essential to proper functioning,
are evaluated for conformance to design input requirements
Design outputs will vary based on the product, technology, discipline and applicable standards.
The regulation and standards are not specific about the content of the design outputs themselves. The significant activity then from a design control perspective is ensuring a trace to design inputs and visibility into the verification of those design inputs.
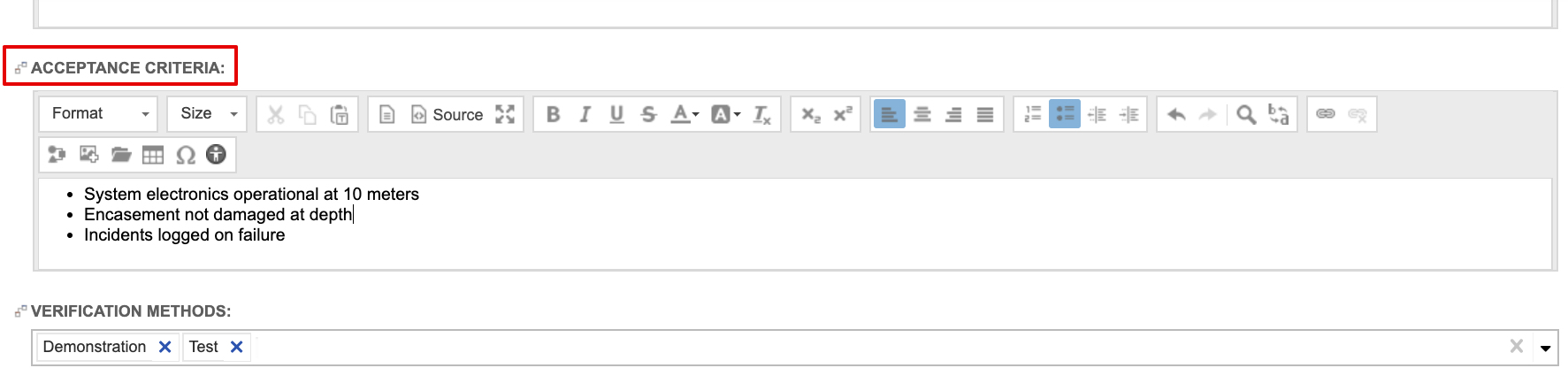
When defining design inputs (i.e., system and subsystem requirements), the guide recommends stating acceptance criteria directly in the requirement items. In fact, the system and subsystem requirements are configured capture it out of the box. Below is a sample System Requirement using the out of the box system requirement configuration.
Verification items, traced to design inputs, should be defined based on the acceptance criteria. Similarly, design output items (e.g., architecture diagrams) are traced to design inputs and refer to requirement details and acceptance criteria for design and development.
For some disciplines, like Software, Jama Connect may contain more detailed levels of abstraction and, therefore, may have items that tie directly into development tools. For other disciplines, Jama Connect may contain very basic information and pointers to other systems managing design outputs details, like parts for manufacturing.
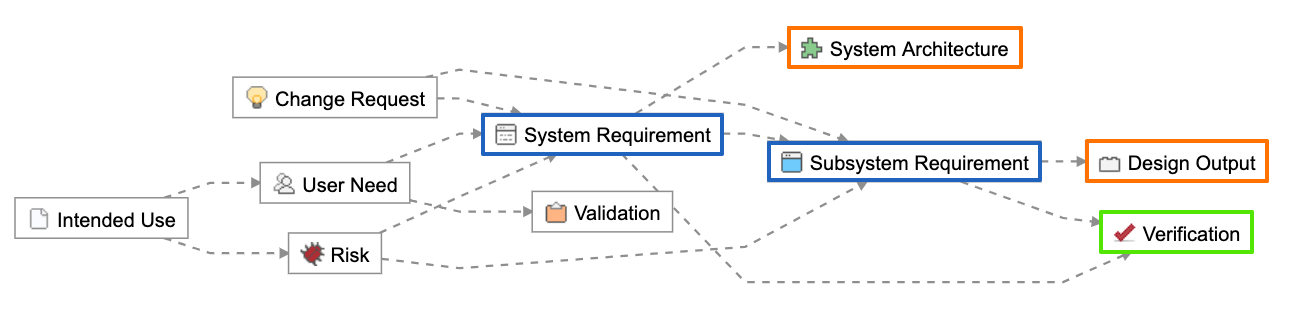
The Relationship Rule below provided with the Jama Connect for Medical Device Development out of the box configuration shows how design outputs (in orange) and verifications (in green) trace directly to design inputs (in blue).
Using this approach, the design input serves as the point connecting design outputs and verifications. Design outputs contain or point to evidence of the implementation of the design input, while verifications provide evidence through testing that the intent of the design input is met. Both design outputs and verifications reference the acceptance criteria captured in the design input during their definition and by nature of the trace.
Connecting design inputs with design outputs and verifications.
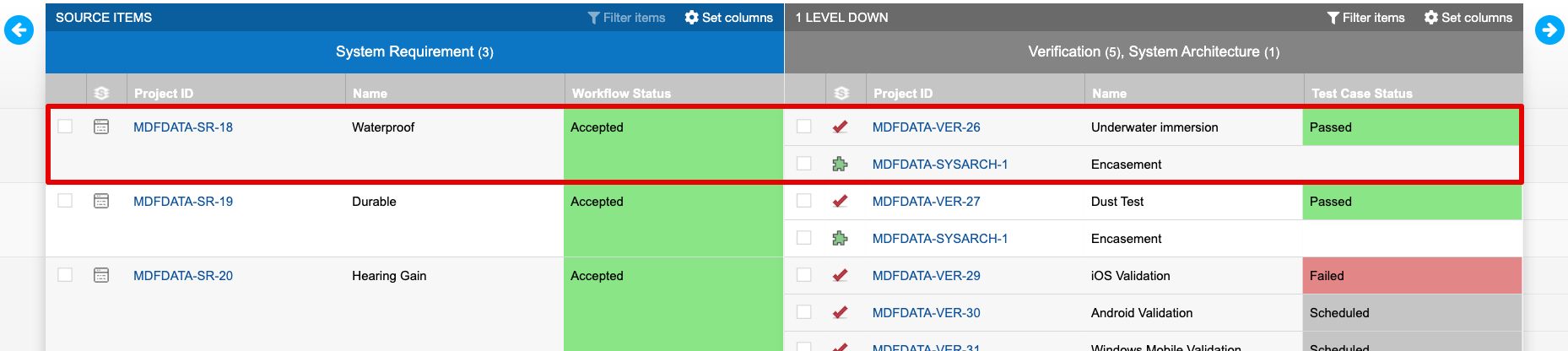
The trace established by connecting design inputs with design outputs and verifications is visible in Jama Connect’s Trace View. The sample of a Trace View below shows a requirement with two related items downstream, a verification and design output. The verification in this example shows “Passed”, which indicates the intent of the requirement was met via implementation of the related design output.
By capturing acceptance criteria during the Design Input process, exposing that information to related downstream processes, and using trace as a part of normal product definition activities, manufacturers gain visibility into the conformity of design outputs and build confidence in their verification activities and results. As seen in the Trace View above, relating information in Jama Connect is key to understanding coverage and verification.
In the next blog in this series, we’ll explore making further use of the trace, beyond simply matrix generation.
Haven’t read the earlier blogs in the series? You can catch up here.
Watch a demo to see Jama Connect Medical Device Development Solution features in action and understand how teams use it to support their development process.
Medical devices have become increasingly complex, connected, and integrated systems. Yet many engineering teams still rely on documents and spreadsheets to manage development, risk assessments, and testing of devices to provide evidence of design control. This approach is no longer viable.
• Why documents-based requirements processes are no longer sustainable
• How to align people, process & technology to improve agility and flexibility
• How collaboration streamlines the development process and improves quality
• How to accelerate NPI of complex, connected devices to anticipate market demand
• How to align validation & verification to meet global regulatory requirements
• Case studies from companies using an integrated platform to improve outcomes
Register and you can watch the on-demand recording now.
As medical device manufacturers develop complex products, they require a product development approach capable of managing that complexity.
At the same time, manufacturers must continue to ensure compliance and alignment with regulations and standards. These define requirements that ensure safety and quality and reduce risk—but ultimately do not prescribe specific tools or techniques.
This is especially apparent in design control activities. Regulatory requirements define the “what” for compliance but leave the “how” to the manufacturer, as long the procedures describing that “how” remain documented and prove sufficient as part of the quality system.
This lack of a prescribed approach to managing design controls can lead to uncertainty, but Jama Connect for Medical Device Development envisions that gap as an opportunity. Here, manufacturers have the space to deploy systems engineering techniques within design control activities, supported with a robust requirements, risk, and test solution.
Note: Now that our medical device blog series is concluded, you can go back and read the series intro, Part I, and Part II.
Bring systems engineering into the design control process to manage medical device complexity.
Systems Engineering solves the problem medical device manufacturers face. It takes a complex problem and divides it into manageable units so developers can see the solution both holistically and in its interrelated parts.
Aligning to 21 CFR 820.30 and ISO 13485:2016 can naturally guide manufacturers towards this approach, requiring trace across user needs, design inputs, design outputs and verifications. The guidance does not account for the abstraction required within these areas, especially design inputs, allowing the manufacturer to determine and implement appropriate techniques and tools.
The Jama Connect for Medical Device Development solution includes:
A Procedure and Configuration Guide
An out-of-the box configuration of Jama Connect
Together, these components bring Systems Engineering principles and design control requirements into a single recommended approach.
Here’s how.
1. Apply systems thinking.
The FDA guidance (FDA, 1997) indicates that product concepts are to be “elaborated, expanded, and transformed into a complete set of design input requirements.” Jama for Medical Device Development’s procedure guide applies systems engineering principles during this design input process.
User needs are fulfilled by functions of the system.
The system allocates them to specific engineering teams or product components for further, discipline-specific definition.
In some instances, like where software is the system, abstraction of design inputs from system-level to subsystems may not cross disciplines. However, the need for hierarchical product definition remains and is reinforced by software specific standards such as IEC 62304 and IEC 82304.
2. Capture and organize design inputs.
In Jama Connect, these levels of abstractions are managed as Item Types, discrete objects within Jama Connect that allow the solution to enforce rules for how information should trace to one another.
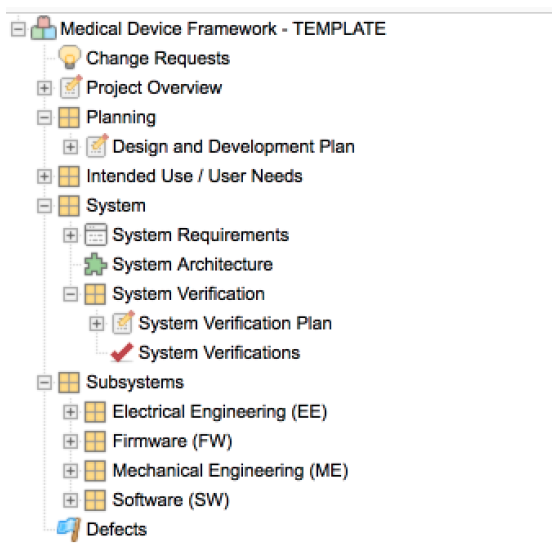
Concept-level information is captured as Intended Use and User Needs, engineering design responses as System and Subsystem Requirements. The total resulting set of requirements are referred to as the Design Inputs.
3. Establish the trace.
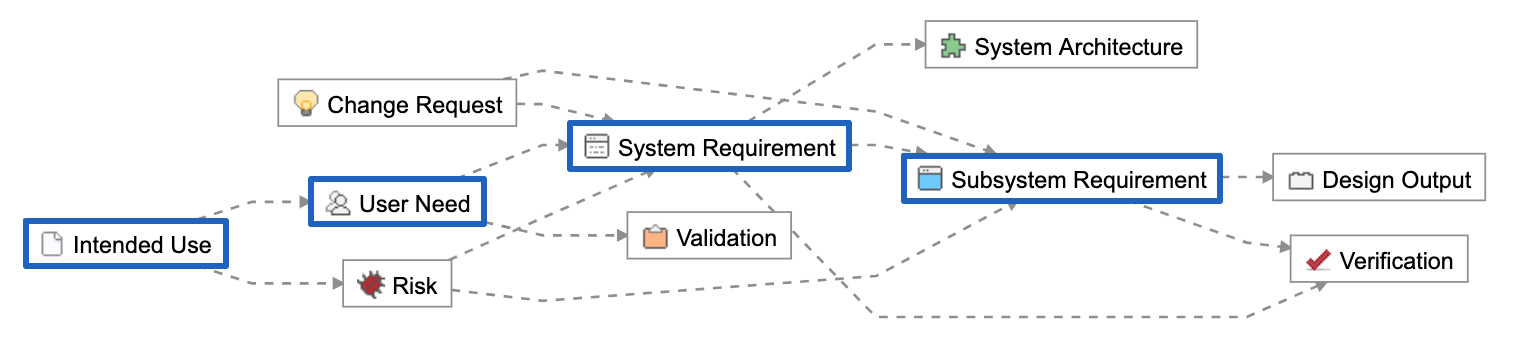
Below is a Relationship Rule provided as part of the out-of-the-box configuration of Jama Connect for Medical Device Development. In blue are the concept-level and design inputs:
These Item Types are unique data elements within Jama Connect. They allow us to create logical groupings we can use to manage the hierarchical levels of abstraction and to further organize across disciplines.
Standardizing engineering principles for requirements management using discrete Item Types is a valuable shift. It shows customers shifting focus from pure document generation to support for how they actually work, which can range from top-down, to middle-out, to bottom-up product definition.
The end result: Actionable information and accepted design inputs.
This shift to a focus on discrete items instead of whole documents encourages teams to act on information as it becomes ready. By capturing status within each individual item (e.g., a specific system requirement), items are independently driven from “Draft “to “Accepted.” “Accepted” indicates a requirement is ready for:
Documentation in the Quality Management System (QMS).
Further decomposition or development.
Defining Verification.
This single state can initiate several activities for other teams and does not require full document completion, which tends to restrict visibility and reduce momentum.
Although this approach encourages a shift from a document-focused product definition, in reality a document needs to be generated for the Design History File (DHF). To support these potentially conflicting needs, the out-of-the-box configuration:
Drives organization of information based on a systems engineering approach.
Uses Jama Connect’s filtering capabilities to pull together information across the project for document generation.
Using filters for document generation allows you to do more than collect different types of requirements for a single document. You can use data within the items, specifically in the Status of the items, to generate a document of only accepted design inputs. You can also take a baseline as part of the document generation process. The result is a snapshot of accepted design inputs that you can use for comparison reporting to show how design inputs may have changed over time. You can also indicate in each item’s version history which version of a requirement was included in the generated documents.
Use this Design Inputs approach with the Jama Connect for Medical Device solution to make it easier to generate documentation you need while supporting how you work with systems engineering approaches.
In the next post in this series, I’ll show you how to connect design inputs with design outputs and verifications.
Watch a demo to see key Jama Connect Medical Device Development Solution features in action and understand how teams use it to support their development process.
We wanted the solution to offer a collection of training and documentation components that aligns to industry regulations so product development teams could get ramped up quickly.
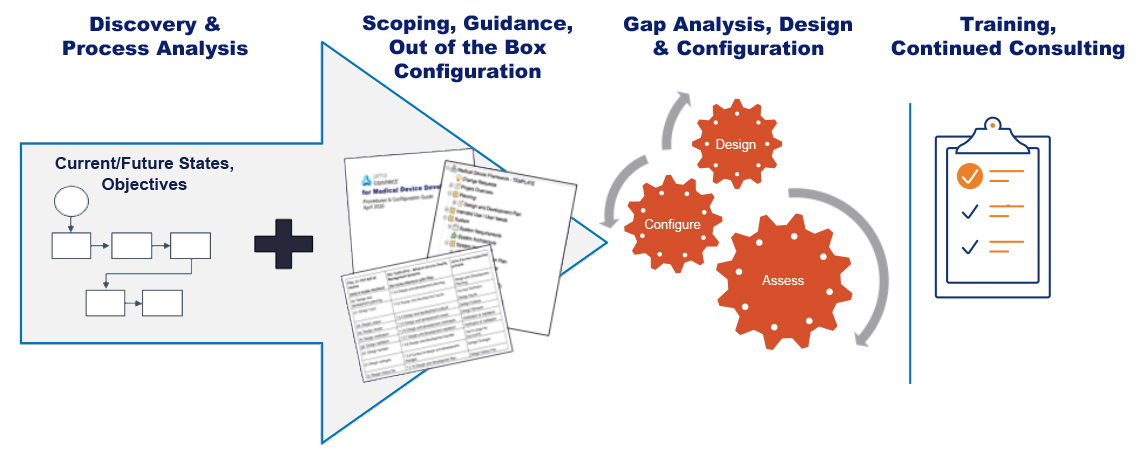
The result: an out-of-the-box configuration of Jama Connect, an accompanying Procedure Guide, and Jama Professional Services. It’s a solution designed to reduce time-to-value, account for and provide tailored guidance around customer-specific needs, and drive adoption through tailored training and on-going support. Let’s take a look at the components and see how they help achieve those goals.
Note: Now that our medical device blog series is concluded, you can go back and read the series intro, Part I, and Part III.
Procedure and Configuration guide: Jama Connect, clearly aligned with regulations.
The Jama Connect for Medical Device Development Procedure and Configuration Guide provides detailed alignment between these regulations and standards and the out of the box configuration and recommended use of Jama Connect:
ISO 13485:2016
ISO 14971:2019
21 CFR 820.30
The scope and processes described in the guide are clearly identified and justified.
An important point to note: we identified the Jama Connect capabilities that align with the most relevant design control requirements needs. Then we targeted those needs, and optimized Jama Connect to meet them exactly.
The regulations and standards establish the design control requirements and provide some guidance, but they are not prescriptive in terms of tools or techniques. The procedure and configuration guide explains in detail how to bring in best practices from systems thinking, supported by use of Jama Connect, while still complying with design control requirements. .
Teams building complex medical devices will benefit from this systems-thinking approach and the way the Procedure and Configuration Guide builds these layers into the configuration. The principles and guidance provided by the Systems Engineering Body of Knowledge (SEBoK) and the recommended use of Jama Connect will bring clarity around product definition and verification activities.
Out-of-the-box-configuration: Start with standardized guidance and build from there.
The out-of-the-box configuration provides a recommended project template that allows customers to get up and running quickly. It also serves as a starting point for customer-specific discovery and configuration. Each aspect of the configuration aligns with the applicable regulations and standard through detailed process activities and tasks described in the procedure guide
The configuration also provides Export Templates you can use to generate documentation from Jama Connect for transport into your Quality Management System (QMS), typically used to house all Design History Files for sign-off and audit.
On-site consulting and training services to align your teams quickly.
Jama Software’s Professional Services team works to help customers reach several goals:
Build adoption, decrease time-to-value, and maintain ease of use.
Focus on process alignment, people readiness, and best practices for configuration and use.
Take advantage of the recommendations and guidance described in the Procedure and Configuration Guide and built into the out of the box configuration.
Provide end-user training tailored to the customer’s use and configuration of Jama Connect .
Taken as a whole, the Jama Connect for Medical Device Development solution provides the guidance, justification and a starting framework that not only answers “what” Medical Device manufacturers should do when deploying Jama Connect, but also “how” and “why” they should do it.
In upcoming blogs in this series, we’ll explore specific aspects of the Procedure and Configuration Guide and the out of the box configuration of Jama Connect. We’ll also look at how the solution components described in this blog are applied in practice.
Download a Solution Brief for an overview that explains all the ways Jama Connect for Medical Device Development can help your teams manage shifting complexities and maintain product quality and safety.
Today, Jama Software announces a new solution: Jama Connect for Medical Device Development. The launch feels especially relevant right now. Over the next few weeks I’ll share details about it with you here in the blog. I’m going to talk about:
The solution background: why it came to be and what it does.
Solution Components
Design Inputs
Connect Design Inputs, Design Outputs and Verification
Traceability
We’re pleased we’ll be able to help medical device manufacturers support design control activities in the highly regulated product delivery environment and contribute to their goals of quality and safety.
Note: Now that our medical device blog series is concluded, you can go back and read the series intro, Part II, and Part III.
Streamlined and focused configuration for medical device development.
The idea for the Jama Connect for Medical Device Development solution grew from the need to align quickly to medical device regulations and standards within a requirements platform.
The highly configurable Jama Connect platform allows customers to align the system with specific requirements, risk, and test process needs. If there’s a particular type of information to capture or a specific list of values that would support the overarching process, customers can configure Jama Connect to align and support it.
However, a highly configurable system introduces challenges. During deployments, you have to ask, “Yes, we can do anything, but what should we do?” Think of a highly configurable system as a blank canvas. You can find yourself staring, directionless, and not know where to begin.
That matters when there’s an entire team staring at the blank canvas. It wastes resources and impacts the time it takes to gain value from the system. In the end, the result can be a misaligned, frustrating deployment and low user adoption.
An out-of-box configuration that brings immediate benefits.
The challenge described above is precisely why Jama Software created the Jama Connect for Medical Device Development solution. We wanted to help Medical Device manufacturers who deploy Jama Connect in a highly regulated environment:
Increase the value of Jama Connect, and decrease the time to realize that value.
Access a recommended, out-of-the-box configuration.
Maintain alignment with design control regulations and standards that Jama Connect is best suited to address.
With the Jama Connect for Medical Device Development solution, deployments no longer start with a blank canvas. Instead, the picture comes out of the box, justified and properly aligned with medical device regulations and standards. Configuration is still possible and valuable, but streamlined and focused.
Customers can hold an out-of-the-box configuration and recommended guidance against their operating procedures to:
Identify and justify unique, customer-specific needs.
Determine the best approach to address those needs.
Incorporate customer-specific nuances for tighter alignment to the company operating procedures.
In next week’s blog post, we’ll explore solution components in more detail.
Discover more details and watch a demo of the new Jama Connect for Medical Device Development solution here.
Today, Jama Software, introduced Jama Connect for Medical Device Development,a new solution designed to help engineering teams better manage device requirements, risk and design control all on one powerful platform. The newest release makes regulatory submissions and audit preparations a straightforward process, speeding time-to-market deployment with no reduction in quality.
Consistent product quality and safety proves to be the top challenge for innovators throughout the medical device development lifecycle. Constantly evolving regulations and the new complexities presented by connected devices makes management of these shifting complexities difficult. Our teams have been hard at work to bring this solution to fruition and help medical device developers simplify and accelerate the development lifecycle. This single platform can help accelerate innovation, maintain consistency and high product quality, and manage ever-changing complex regulations in medical device development.
“Developers are balancing increased regulatory requirements with device development costs that lead to challenges across the production lifecycle,” said Josh Turpen, Chief Product Officer at Jama Software. “We’re excited to introduce our new solution designed specifically for medical device companies which will help ease the development process from the start. Jama Connect allows developers to hit the ground running with preconfigured templates and best practices built-in, saving product teams from the cumbersome and often frustrating process of filing paperwork.”
Note: Now that our medical device blog series is concluded, you can go back and read Part I, Part II, and Part III.
Jama Connect for Medical Device Development can help teams:
• Easily demonstrate traceability
• Manage risk analysis
• Maintain audit trails and export data
• Improve product development with reuse and baseline management
• Streamline compliant reviews and approvals
• Manage design verification and validation
Jama Connect for Medical Device Development is designed to help you get ramped up quickly with a platform, training and documentation aligned to industry regulations ISO 13485:2016, 21 CFR 820.30, and ISO 14971:2019, while applying a proven Systems Engineering approach to product development. When you purchase Jama Connect for Medical Device Development, our consultants partner with you to adapt the solution to fit your product delivery process and build adoption of Jama Connect within your organization. Contact us to learn more.
Jama Software provides the leading platform for requirements, risk and test management. With Jama Connect for Medical Device Development, teams building complex products, systems and software improve cycle times, increase quality, reduce rework and minimize effort proving compliance. Representing the forefront of modern development, Jama’s growing customer base
of more than 600 organizations includes Boston Scientific, Johnson & Johnson, Abbott, and Merck.
Editor’s Note: In a time where remote collaboration and distributed teams are quickly becoming the norm rather than the exception, we’re proud to share this post on the value of effective collaboration around medical device design and product development. This article was originally published here on May 5th, 2020 by MedTech Intelligence and written by Jama‘s VP of Customer Success, Clay Moore.
How Effective Collaboration Can Expedite Medical Device Design
Medical device companies are committing significant resources to the fight against Covid-19. Collaboration tools can help expedite product design while supporting evolving compliance standards.
Weeks after the COVID-19 lockdown, medical device companies continue to face challenges as they manage remote engineering teams working rapidly to keep up with a changing environment. Now that the initial shock has passed, companies are becoming more well-versed in their new workplaces and identifying how to keep remote engineering teams working successfully.
Teams are under pressure to meet quality and compliance standards while staying on pace to hit delivery dates, and optimizing the efficiency of remote engineering teams as they develop complex devices can be difficult. Normally, products can undergo multi-month-long review cycles, which tie up valuable resources. Technology, when properly leveraged, can reduce that review cycle by as much as 75%—a vast improvement.1
Effectively streamlining collaboration is key to expedite design reviews and medical device product launches. The following is how companies can use collaborative technology to maximize output while meeting critical health standards.
Reduce Dependency on Documents
Medical device companies have been forced to come to terms with their current processes to see if they work for remote engineers. Most know that helpful technology exists, but platforms that promise to streamline lengthy processes can be intimidating. Companies that delayed digital adoption are especially disadvantaged.
The document-based requirements management approach often used by medical device companies limits visibility into the design process across teams. This makes it difficult to scale across multiple product lines and versions, especially with teams working remotely, and it increases expenses.
Collaborative requirements management software can help engineers easily communicate and smoothly define, review and validate information digitally to ensure projects are tracking and a clear path to compliance and launch is visible. Effective software reduces manual processes that limit innovation and add time to development.
Medical device companies should seek collaboration tools that help their engineers clearly communicate the path to launch, tasks and ownership, and current status and action items needed. Remote work on complex processes requires seamless communication and understanding where work is being bottlenecked so teams know how to direct their attention.
Stay On Top of Compliance
In the medical device industry, understanding current product regulations is key to shape the product development process. In recent weeks, the FDA has issued changes to its 510(k) program, and the European Parliament voted to delay the EU Medical Device Regulation.2,3 Companies who have a centralized approach and real-time access to requirements and design can minimize the added overhead of regulatory requirements, streamline development, and minimize risk.
When companies lack resources to efficiently locate regulatory standards and swiftly communicate them to engineers, they’re wasting time and limiting the ability to mitigate risk. Using traditional collaboration tools or Word documents to manage the product development process isn’t effective, leading to teams spending days or weeks to pull together documentation in preparation for an audit. Ensuring compliance requires context and visibility throughout the design process, and innovative tools that structure collaboration and put compliance and regulation at the forefront can provide both.
Good requirements management software uses a lifecycle approach to make sure compliance is integrated into the design process. Team members are held accountable, and workstreams keep track of when signoffs occurred.
Getting compliance right can be a challenge with engineers working remotely—but it is too important and too costly to get wrong. It can cost $600 million to recall a product.4
Remote Collaboration Sets Medical Device Companies Up for Success
Companies forced to embark on their digital transformation journey during this pandemic can save valuable time and money on product launches by embracing requirements management software to help streamline communication, structure collaboration, and ensure compliance.
This level of collaboration is more prevalent than ever, especially considering the state of work may change as we know it. In fact, a Gartner, Inc. survey revealed that 74% of CFOs and finance leaders will move at least five percent of their previously on-site workforce to permanently remote positions post-pandemic.5 Engineering teams need to make remote collaboration an effective part of their normal operating routines, starting now.
By embracing the right technologies, medical device companies are not only investing in supporting their teams at this crucial time but also they are investing in the success of taking products to market, as the world conducts more and more business online.
Take a look at some of our other resources around collaboration that we’ve compiled for easy review here: READ MORE
What Phases are Needed for Developing a Medical Device?
Developing a medical device is an inherently complex process, and one that’s becoming more complicated all the time. In addition to the increasingly stringent regulatory requirements that must be met for FDA approval and/or EU MDR compliance, medical device manufacturers must also navigate ongoing changes across the industry, including the rise of connected devices within the Internet of Things (IoT).
Making it all the way from initial device discovery and conceptualization through to FDA clearance and launch strategy requires a dedicated team, as well as a risk and requirements management strategy supported by modern tools. Without such solutions in place, the high costs and significant risks of developing a modern medical device become more challenging:
The average cost of development under the Food and Drug Administration’s Premarket Approval (PMA) program is $94 million. For the less strict 510(k) path, it’s still a considerable sum – $31 million.
Product recalls are more expensive. A McKinsey study estimated the cost of these events at as much as $600 million apiece. Indirect losses from reduced market cap and lost revenue can run into the billions of dollars.
Medical device security is also paramount in the age of the IoT. Internet connectivity can expose devices to cyberattacks and the healthcare industry is routinely among the most targeted sectors.
To fully account for the relevant challenges and also zero in on corresponding solutions, let’s look at the process a medical device must go through before it reaches patients.
The 5 phases of developing a medical device
Medical device development is a multi-phase process, typically with five main stages. A sample process – this one outlined by the FDA itself – proceeds as follows:
Phase 1: Device Discovery and Concept
There are several overarching questions to answer at this initial stage:
Is there an unmet need that a new device could potentially satisfy?
What risks would the device, if brought to market, pose to patients?
How might those risks translate into the device’s classification?
Can a proof of concept outline a workable path to regulatory approval?
The classification of the potential device, in particular, will greatly affect the product development process and necessary risk management procedures.
For example, Class I devices are subject only to general controls such as good manufacturing practices and recordkeeping requirements, while higher-risk Class II and Class III devices require additional special controls and PMA, respectively. Accordingly, risk management is more complex for Class II and Class III devices, plus the associated costs are higher.
Phase 2: Preclinical Research and Prototyping
At this point, the device isn’t suitable for human use yet, but it is ready for testing within controlled laboratory environments. Observing the prototype’s performance under these conditions provides some early insights on how it might be used by people, and into what specific risks might accompany it.
Phase 3: Pathway to Approval
The device’s classification/risk level will determine the subsequent steps.
A Class I device such as an oxygen mask has to comply only with general controls, and many devices in this low-risk class are exempt from any premarket submissions. Higher-risk Class II devices, such as pregnancy test kits, must adhere to special controls, like meeting device-specific performance criteria, on top of those general controls.
Non-exempt Class I and Class II devices require a 510(k) Premarket Notification proving “substantial equivalence” with a legally marketed device that isn’t subject to PMA. If such equivalence cannot be proven, the device is placed into Class III.
Class III devices – including those nonequivalents, along with devices that sustain or support life – require PMA, which is the most complex part of developing a medical device. Scientific evidence must be submitted showing that the device’s benefits outweigh its risks and that it will help a significant portion of the population.
Phase 4: Regulatory Review
Once a medical device company has sufficient data on how its device performs and the accompanying risks, it can apply for regulatory review. The exact process will vary by device classification/risk level.
PMA applications entail a thorough review of the laboratories and facilities for production for good manufacturing practices, and an evaluation of the results of related clinical and nonclinical studies. Other processes are simpler.
Phase 5: Post-Market Device Safety Monitoring
After the device reaches the market, it will be continually monitored for possible safety and performance issues. Regulatory bodies may inspect manufacturing facilities again, while consumers and medical professionals can report any observed issues through programs like MedWatch in the U.S. This would be the stage at which a recall would be initiated, if applicable.
Throughout all five phases, development teams will need to maintain many different documents, including design history files for each finished device. To ensure that their work stays on track and complies with key regulatory standards such as ISO 13485, ISO 1497, and EU MDR provisions, it makes sense to create a single source of truth that supports scalable, efficient requirements management.
Upgrading requirements management for modern medical device development
Providing a medical device’s compliance can be complex. Traditional document-driven models are not ideal for this purpose, either, as they can involve many discrete spreadsheets and other files that take a long time to retrieve, review, and organize. The inefficiency of these workflows also complicates the traceability of development activities back to requirements.
In contrast, a centralized platform, with all key information in one place, can provide clearer insight into design controls for device requirements and related risks. Teams can use Jama Connect to gain real-time visibility into how design inputs have been met and verified, providing necessary evidence from the design control process, for better-informed decision-making and a more streamlined overall development process.
Defects can also be identified earlier and more reliably, curbing the risk of noncompliance and recalls. To learn more about we can help support your medical device projects, get in touch with a Jama Connect expert today.
If you’d like to learn more about medical device development,
we’ve compiled some helpful resources all in one spot! LEARN MORE
Building and then bringing a medical device to market as quickly as possible—all while preserving acceptable levels of quality and regulatory compliance—requires adept medical device risk management. By minimizing potential risks such as mislabeling and software-related issues, medical device manufacturers make each product safer for the patients who will use them. All of these risks and others are present through the product development lifecycle, where they must be addressed through specific risk management activities
The ISO 14971 standard, as encapsulated in ISO 14971:2007 and then revised in ISO 14971:2019, is the modern framework for such efforts. An FDA Recognized Consensus Standard, it has a nine-part structure defining the criteria for medical device risk management during production and post-production. ISO 14971 is also required by higher-level regulation under ISO 13485. All medical device companies follow ISO 14971, but their individual approaches to the risk management standard will vary based on not only product type but the actual tools used for handling risk analysis and control measures as well.
Risks as requirements: What’s the best approach to medical device risk management?
Effective medical device risk management is integral to patient health and safety. A study published by The British Medical Journal found that one in 20 patients experiences preventable harm when receiving medical care. Moreover, many medical devices in active use are many years old, meaning that even flaws implemented long ago can continue to pose risks to patients. That means risk must be curbed at every stage of the product lifecycle.
Another way to look at it: Risks are central requirements when it comes to the medical product development process, and safely managing them in accordance with ISO 14971 requires a comprehensive modern solution capable of delivering the necessary coverage, speed and preparation. Risk management is requirements management in the medical device industry. Accordingly, it’s crucial to have the capacity to, for instance, connect eventual verification tests back to requirements, so that teams can be confident of adequate risk mitigation.
However, many existing workflows and tools cannot consistently ensure acceptable compliance, leading to the possibility of recalls, or inadequate workarounds such as alterations to the label or instructions-for-use. Spreadsheets exemplify the limitations of older approaches to requirements management and risk management during the medical device lifecycle.
The problem with document-based processes
Medical device manufacturers may rely on Microsoft Excel to capture risk data and fuel their risk management planning and reporting activities. The potential problems with this approach include:
Limited scalability to teams working at multiple locations.
Siloed data sources that take time to comb through and reconcile.
Human factors such as miskeyed entries or inadvertent deletions.
Difficulty proving compliance, due to lack of end-to-end traceability.
Taken together, these issues make it onerous to maintain and execute on a medical device risk management plan that fulfills all provisions of ISO 14971. This standard requires a combination of risk analysis, evaluation and control – all processes that a risk management plan helps simplify by documenting all of the potential risks across the product lifecycle.
How to more reliably put a risk management plan into action
The risk management planning process should produce a plan that contains product-specific data and follows all standard operating procedures in the domain. The plan should also be a living document that can be continually updated as requirements and risks evolve, as it will serve as the blueprint for ongoing risk management activities such as reporting on hazards and risk control measures and also linking back to requirements. Ensuring acceptable levels of detail and accuracy in the risk management plan is much easier with an all-in-one solution than with a collection of discrete documents.
Let’s say a hypothetical medical device company was developing an MRI machine. If it were centering its risk management processes within a massive Excel sheet, lots of valuable time would be lost to stakeholders on the development team having to constantly review the requirements in the shared asset.
Plus, this highly manual, error-prone process can itself create further complications for overall medical device risk management, such as a risk going initially overlooked due to outdated data in a spreadsheet cell. Going back later to write a report about the risks is not a great alternative to building in risk management throughout the development process—but the right tools are needed for the latter strategy.
By switching to a more modern solution, this development team could instead:
Take advantage of risk plan templates to ramp up more quickly.
See live risk mitigation data, not outdated entries.
Avoid the various administrative risks of Excel, like splitting/merging cells.
Easily adjust probabilities and severities of the defined risks.
Enable real-time collaboration between teams.
Visualize and trace risks across the whole product development lifecycle.
Prove ISO 14971 and other regulatory compliance more easily.
At the end of the development process, the team making this hypothetical MRI machine would be able to see clearly how its verification tests traced back to the risks and requirements it initially set. More specifically, they would know if the product could move the right amount of air, survive the expected transport and storage conditions and comply with all applicable rules and regulations.
Demonstrating ISO 14971 compliance with Jama Connect
Jama Connect offers a modern alternative to document-oriented processes for medical device risk management. Jama Connect is built to help streamline compliance with ISO 14971.
For example, Clause 7 of ISO 14971 requires attention to residual risks, or those risks that exist even after all risk control measures have been implemented. In Jama Connect, those measures can be efficiently defined and linked to corresponding risks for maximum traceability. That way, teams can spot potentially unacceptable risks early on in development and mitigate them before the associated costs and logistics become impractical.
There are many other features within Jama Connect for complying with all clauses of ISO 14971 and modernizing your general approach to medical device risk management to keep up with changes such as the FDA’s Safety and Performance Based Pathway. To learn more, connect with an expert today.
To learn more on the topic of risk management, we’ve compiled some helpful resources for you.