In addition to a walkthrough of the platform and our Live Traceability dashboard, we’ll cover:
The critical challenges to reducing risk in product development
Why deeming requirements “good enough” to allow teams to proceed with an acceptable level of risk culminates in static requirements, unplanned rework, and compounded product risk
How “Project management” activity is a fallacy — it is the management of requirements, people, risks, change, opportunities, expectations, resources, commitment, and suppliers
Below is an abbreviated transcript and a recording of our webinar.
Reducing Risk in Product Development
Kemi Lewis: Today’s agenda covers a deep dive into the critical challenges to reduce the risk in product development, what are the viable solutions to this problem, key takeaways, and wrapping up with a question and answer session at the end of the webinar
Let’s get right into it. What are the main critical challenges that product development teams are facing? In my experience, the main factors that lead to adverse product outcomes and risk are, number one, no upfront and iterative collaboration during requirements and design creation and review stages due to limited customer and cross-functional team involvement in the review and approval of requirements. This lack of cooperation results in missed and misunderstood requirements driving the product design into severely costly errors later on.
Second factor, no digital thread connecting the product and team to the end to end product life cycle process. What do I mean by the digital thread? A digital thread is a data driven architecture that links together information generated from across the product life cycle and is envisioned to be the primary and authoritative data and communication platform for a company’s product at any instance in time. Without this digital thread, there’s no ability to track the life of a requirement through development, test and release.
Kemi Lewis: This missing digital thread results in static requirement documents rarely viewed by critical stakeholders maintained in Word, Excel or standalone tool used only by a few as a repository. I’ve personally experienced this at companies where only the systems engineers were accessing the repository and the rest of the product development team from product managers down to testing and integration engineers never accessed it.
You can only imagine how this turned out. Countless rework during testing and integration in addition to postlaunch rework this early, which was severely costly to the customer and left them very unhappy. So lacking this digital thread leads to no management visibility into crucial metrics for the end to end process and no identification of process risk patterns, such as delays in development, multiple test failures, rework cycles, etc.
Third main factor is having a low level of requirements management maturity. Let’s discuss this in more detail. Level zero: There are no formal requirements. So no documentation exists for user or system requirements. Instead, development operates off of user stories with no clear distinction between the functionality of the system being built and expected user experience. Level one: Document based requirements. Static requirement documents are created and most often maintained by each author on their desktop with various emails, slack comments containing more information. This especially gets fun when you have to merge 10 different versions of the same document from 10 different people from 10 different timeframes, none of which have visibility to each other’s feedback in real time. I’ve seen this at several companies where they lose technical product proposals due to this inefficiency of being able to get a proposal out in time representing the right design specifications of their product.
Kemi Lewis: Level two: Siloed requirements tool. A standalone tool in place to draft review, track comments, version and store static requirements documents, compliance steps, limited reuse, defects and recalls. Level three: System based compliance. Compliance is the forcing function to shift from static to live traceability to meet standards for requirement validation, verification and traceability into a single end to end system. Level four: Product risk reduction. A process centric focus to reduce the likelihood of all forms of product risk via a system enabled live traceability. This requires detection and alerts for specification and functional changes, process exceptions and test failures with resulting impact analysis. The risks mitigated include failure to meet the needs of the customer, failure to perform specific functions, delays, cost overruns, defects, compliance and regulatory gaps, delays and fines in addition to recalls.
And the last level of maturity, level five: Development process improvement. Moving past compliance and risk into the spirit of standard based on quality management and process control. These stages place focus on measuring, managing and improving the product development process. The unintended result of this fragmented process is that critical function such of requirement, traceability, verification, validation, risk mitigation, product integration and compliance are often fraught with information gaps, defects, delays, reworks, recalls, missed requirements and significant manual effort. This includes all areas of the complex product system and software delivery life cycle that can experience negative outcomes and should be actively managed to reduce the likelihood of appearance, such as performance.
One of the early steps I advise my clients to take when developing their medical device is to determine the class and classifications of their medical device. In conjunction with the complexity of the device, understanding the class and classification sets the foundation for your product development timeline and effort.
This post gives a basic introduction to FDA medical device classes and classifications and the implications for your product development schedule and requirements management.
What are FDA medical device classand classifications?
The FDA established three regulatory classes based on the level of control necessary to assure the safety and effectiveness of the device. Classification is based on the intended use of the device and indications for use, as well as the risk the device poses to patients and users.
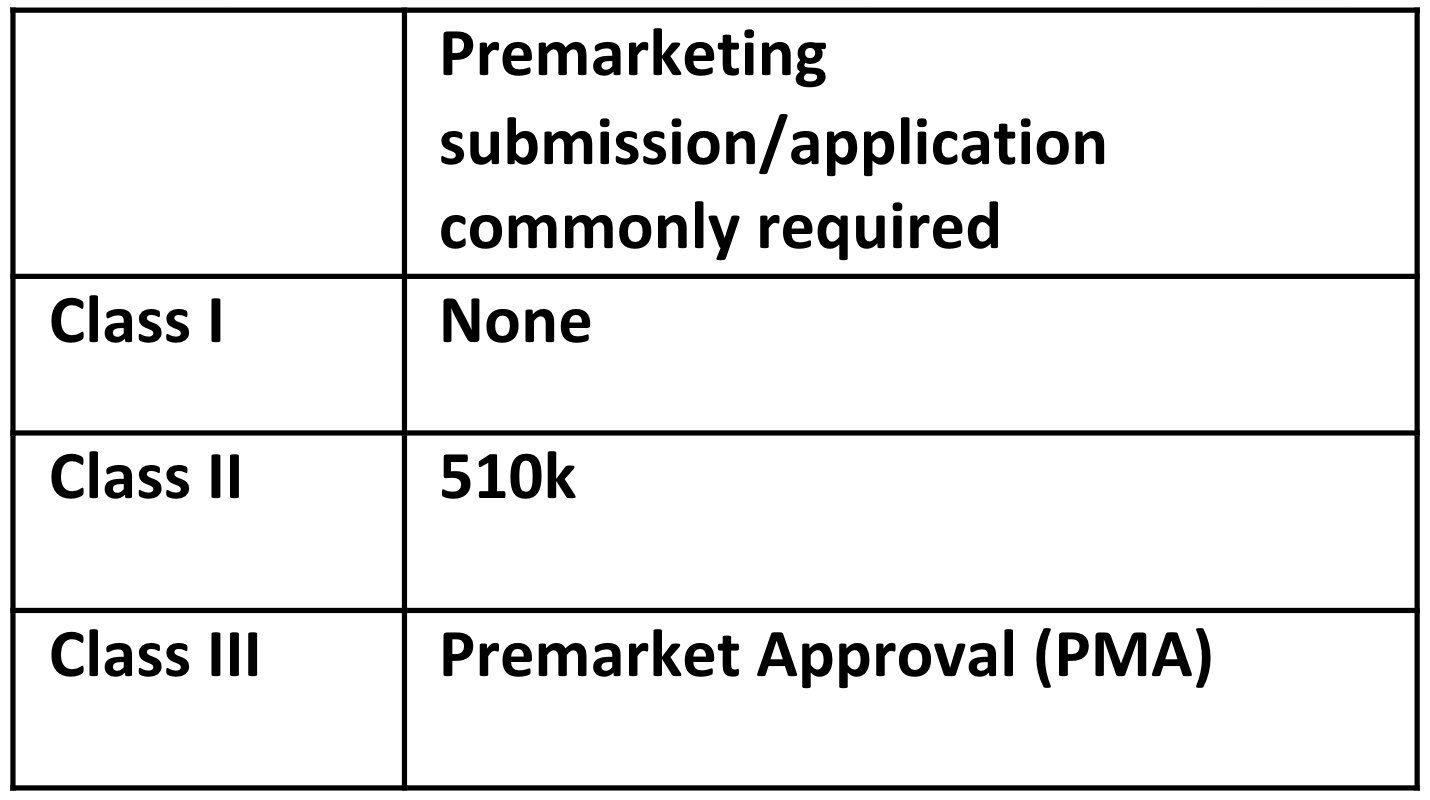
There are three classes: Class I, Class II, and Class III. Class I devices are those with the lowest risk, Class II devices have a greater risk, and Class III includes devices with the greatest risk.
The FDA also established classifications for over 1,700 generic types of medical devices and grouped them into 16 panels, or medical specialties. Example panels include Cardiovascular Devices and Radiology Devices. Each of the generic types is assigned as Class I, Class II, or Class III.
The class and classification of the device impacts what FDA premarket submission or application is required for clearance to market. The common premarketing submission or application for each class are:
Note: These are the common regulatory submission and applications for each class of device. There are exemptions, limitations on those exemptions, special controls that may apply, and exceptions, so be aware whether any of these applies to your device. For example, about a quarter of Class I devices are not exempt, and a 510k premarket submission is required.
As the process for the 510k submission is 30-90 days, and the process for the more in-depth PMA submission is 180 days to accept or reject, this time should be understood and planned into your product development schedule.
Similarly, expect elements from the required design control process and design history file to be included as part of a 510k and PMA. Also keep in mind that when design controls are required for your device classification, the full design history file can be scrutinized as part of an FDA inspection of your organization. Since the FDA evaluates whether a device is effective and ensures the risk to the patients and users is appropriately addressed, good requirements and risk management is key. It’s important to have an organized manner in which to demonstrate and document that risk management and user needs are successfully traced through design inputs, design verification, and design validation. A requirements management tool like Jama Connect™ allows for this traceability in an efficient, collaborative, and regulatory-compliant manner.
Understanding your device class and classification is a key step to understanding the path for FDA regulatory clearance and subsequent design control requirements for your medical device development. Knowing those expectations up front will make for a smoother medical device development journey.
Learn more about developing medical devices with Jama Connect!
Medical Device Risk Management
Medical device developers must ensure risk is addressed as a core activity. The ISO 14971 standard, which has been revised three times, provides a proven and flexible framework around which developers can effectively manage the risk of devices for patients and stakeholders. Knowing the standard and applying some aspects, in a reactive way, is not sufficient to create a safe product. Implementing an effectively proactive process and achieving live traceability throughout development, while adhering to the standard will result in a higher quality and safer product fit for market use.
In this blog, I will share how proactive risk management along with Live Traceability™ are two key areas that every medical device developer should focus on when it comes to risk management.
Reactive vs. Proactive
A lot of medical device developers do not prioritize risk evaluation throughout the different design stages, but instead, consider risk a checkbox activity at the end of development. With important prototype deadlines, limited funding, and resource constraints, it’s very easy to make excuses for not running risk evaluations of initial user needs but waiting until entire subsystems have been designed. This may seem like a minor hiccup, but the lack of ongoing risk assessment from an early stage can be very risky for FDA approval, future patients as well as your business’s bottom line and reputation. The later that risk is addressed, the more expensive changes are to incorporate. Delays become the norm as well as budget overruns.
Jama Connect Enables Live Traceability™ Across Your Development Process:Learn more!
Live Traceability™
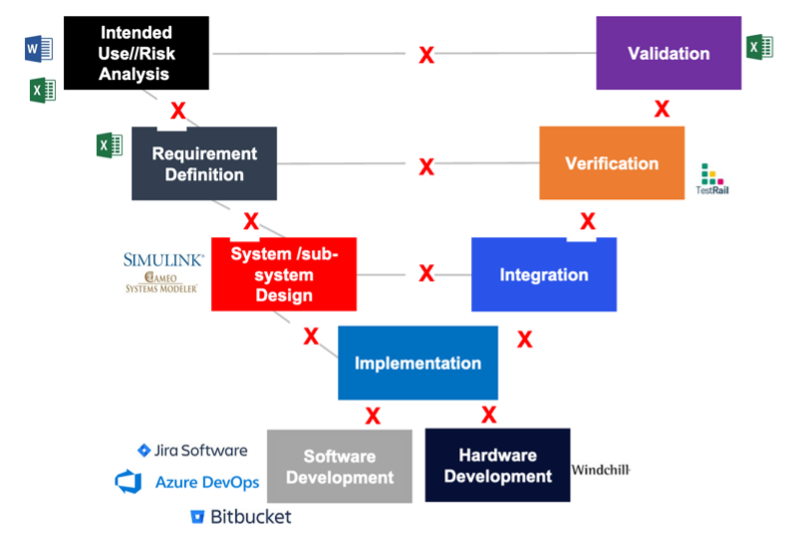
The number one cause of inspectional observations, product recalls & delays, increased CAPAs, and cost overruns is the lack of live traceability throughout different development stages. With complex medical devices, there can be thousands of user needs, product requirements, risks, mitigating requirements, tests being executed, and defects logged. At Jama Software, we’ve noticed that a lot of medical device developers are using different technologies and applications to store this information including Excel, Jira, Word, and others. The below image depicts that and shows the siloed world a lot of engineering teams are living in:
Unless you have a real-time system that can interface with spreadsheets, tools like Jira and other applications, it’s impossible to get a holistic view of development. This can cause several issues, some of the most critical being:
Late identification of defects/coverage gaps due to lack of visibility throughout the development process
Lack of requirement coordination and change management between hardware/software
Lack of ongoing risk assessment and change management
Cumbersome and risky effort to produce trace reports and other DHF artifacts
Without Live Traceability, risk teams can’t be effective in mitigating potential failures or hazards. Outdated and manually updated trace reports and spreadsheets can’t be relied upon. What if there was a way to get a real-time view of traceability throughout development?
At Jama Software, we’ve created the capability for our product development platform to integrate seamlessly with tools like Excel, Jira, and others straight out of the box, and we are proud to be the only vendor on the market that can help engineering teams realize Live Traceability. Risk should be a priority, not a checkbox item!
Get in touch with us to see how we can help you.
This is Part 2 of a series examining the role legacy requirements management solutions, such as IBM® DOORS®, play in introducing project risk to the product development process. To read Part 1, visit Why Migrate, Why Now?: Part 1
5 Common Migration Myths Debunked
Transitioning to a new solution doesn’t have to be challenging; however, there are some assumptions that mislead us into thinking that difficulty is inevitable. Consider the following myths:
MYTH 1: Migrating away from IBM solutions will be more expensive. The amount of work that goes into upgrading to IBM DOORS Next or transitioning to a new RM solution is the same, differentiated only by the quality of the tool and services available to help with migration. An option other than the DOORS family is most often a better fit for your organization.
MYTH 2: Customization will carry over to DOORS Next. You spent a lot of time customizing IBM DOORS and may believe those customizations will transition seamlessly to DOORS Next. However, this isn’t the case and is the reason selecting a different solution doesn’t involve more work.
MYTH 3: DOORS is already deployed and cheap to maintain. Continuing the current path with IBM DOORS is an expensive option in the long term, and often requires dedicated personnel. Switching to an alternative RM solution can improve efficiency while saving money.
MYTH 4: Business disruption is too difficult. The right RM tool will empower teams to effectively hit deadlines, collaborate, and improve business outcomes.
MYTH 5: The user experience will suffer. Many people refuse to use DOORS due to a challenging user experience. DOORS Next is a completely new tool with a new user experience. Adopting a user-friendly solution allows teams to collaborate far more effectively as team members can accelerate concepts, designs, and validations for faster times to market.
The fact is, IBM® DOORS® is extremely outdated, and at some point, updates and support will inevitably end.
If you’re currently using DOORS, you likely know that moving to a different solution is necessary, and you might be considering DOORS Next. However, fast-shifting market dynamics require a new approach to accelerate innovation. As a modern alternative to traditional legacy platforms, Jama Connect® enables digital transformation with a more efficient and user-friendly approach to managing risk and compliance.
Customers agree, naming Jama Connect the overall leader (#1) in requirements management software on G2, outranking IBM DOORS Next for implementation time, adoption, ROI, and market presence.
Jama Connect is a proven IBM DOORS alternative with flexible and reliable solutions, including:
Operation in an IBM DOORS supply chain.“Innovative companies leverage Jama Connect to get up and running fast with a modern requirements management process that tightly aligns with industry standards and practices that support regulatory compliance. Organizations can connect to customers and suppliers that use IBM DOORS through Data Exchange for Jama Connect.”
Integration services. Jama Connect provides integration with key product development lifecycle tools.
Coexists with IBM DOORS. IBM DOORS is embedded in many organizations and may take some time to migrate completely. Progressive teams and divisions can get started on Jama Connect quickly while the larger organization works toward replacing existing programs over time. This approach is supported through a mix of integration, migration, and exchange services.
“Jama Connect lowers the complexity and burden of having to manually keep requirements, architecture and specifications all in sync and traced to each other. It’s a formidable problem that is virtually eliminated courtesy of Jama without the hassle of having to learn a clunky UI (IBM DOORS).” Alan M., Chief Product Officer
Jama Connect® created Live Traceability™ management which reduces project development risk by forming a digital thread through siloed development, test, and risk activities. The flexible platform is designed to support the end-to-end product development process with:
A simple, single repository so it’s easy for remote teams to gather, review and execute on requirements.
Structured reviews and collaboration — teams can elicit feedback, review product features in real-time with stakeholders and track critical decisions across teams and locations.
Change management throughout product development — end-to-end traceability and real-time collaboration improve visibility and make it easier to adapt to changes and track their impact.
Integrations across the ALM-PLM ecosystem.
Moving from IBM® DOORS® Next to Jama Connect® for Requirements Management
To adapt to shifting and ever-increasing challenges and complexities and keep pace with the competition, innovative organizations are now requiring best-in-class software to scale development, reduce risk, save time, and ensure compliance to quality and safety regulations.
Download this paper to learn how Jama Connect stands out above DOORS Next in the G2 Summer 2021 report for requirements management in the following areas:
Products, systems, and software development are only getting more complex and not modernizing your requirements management process will increase the probability of negative outcomes in your product development process.
As your team requires the ability to adapt, innovate, and grow, continuing to use IBM DOORS will become more difficult and will introduce significantly more risk in your product development process. Think about DOORS as a landline rotary phone that stopped being on the receiving end of upgrades after the industry switched to marketing push-button touchtone phones. (And now there are smartphones available, which are mobile and make you even more connected and productive.)
Transitioning to new technology provides your teams with the tools required to innovate, meet deadlines, and succeed. A modern requirements solution can help you to define, manage, and validate complex systems requirements while eliminating the risks and inefficiencies associated with documents and legacy systems.
Hear about the experiences of the more than 50 companies that have made the switch from IBM DOORS to Jama Connect.
Connect with us today to learn how you can enable end-to-end compliance, risk mitigation, and process improvement with our intuitive, award-winning requirements management platform.
Congratulations! Your organization has gained regulatory approval and launched its medical device product. The ‘History’ in Design History File may elicit impressions that all those design and development requirements are now done and considered part of the past. However, several components of the DHF continue as a reference and evolve, including requirements and risk management. Here are 3 ways active management of requirements and risk continues after commercialization:
1: Post-market surveillance
Once your medical device is on the market, post-market surveillance programs, including complaint management processes, must now be exercised. That includes evaluating feedback, determining if it is a complaint, investigating complaints, and determining whether to initiate corrections or corrective actions. As part of this process, requirements and risk management are being used in 2 ways, 1) as a resource to evaluate complaints and 2) a living document to be updated with the experience gained.
As a resource, it is important to reference risk management files to determine if the frequency of occurrence and types of failure modes documented during design and development matches the infield data being gathered. A more frequently occurring failure or new failure mode indicates an investigation is warranted and re-evaluation of the risk. Depending on the outcome, corrective action may be needed.
For example, during design and development, it was determined that a sensor failure leading to customer annoyance occurred rarely, leading to a low risk rating at the time of market launch. The first year on the market, reports of this failure occurred rarely, matching the occurrence rates in the risk management file. Given the low risk and lack of trend, further failure investigation and corrective action were not taken. However, one year later, a change in supplier coincides with a change in occurrence from rarely to frequent, leading to a medium risk. This increase in risk prompts an investigation to determine why the sensor failure rate is higher and to determine corrective actions and controls with the new supplier.
As a living document, the risk management files are to be updated with the observed occurrence rates, new cause(s) of the failure mode of the sensor, mitigations and controls put in place, resulting verifications, and revised risk rating.
2: New Products
Another reason requirements and risk management continue once a product is commercialized is to aid in the development of new products, including line extensions, new models, and next generation platforms and portfolios.
The existing product’s requirements and risk management, supplemented with what is learned from post-market surveillance and other feedback from the field, provide the foundation for new products. A requirements and risk management tool like JAMA Connects® can simplify the management of requirements and risks shared between products to keep teams aligned and prevent requirements or risks being missed during the transfer from one product’s design history file to another. Likewise, line extensions can be more easily incorporated into an existing design history file if requirements and risk management have been properly updated as needed and are accessible.
3: Change Control Evaluation
Change control evaluations is another way management of requirements and risks continue after commercialization. Changes to a product and how it is manufactured occur for many reasons, including replacement of a component that has reached its end of life from a supplier, software upgrades to address bugs, duplication of a manufacturing line, and changes that address complaints.
Changes must be evaluated as to their impact on the form, fit and function of the product, and can have varying degrees of potential impact. Well managed and active requirements and risk management, with traceability to design outputs and verification, become a strong tool for organizations to evaluate the potential impact more quickly.
For example, say a temperature sensor was added as mitigation to prevent overheating of a medical device; overheating that could result in burns to the patient. The sensor, including the necessary accuracy, is listed as a control for the risk of overheating and burns. There’s also a corresponding design requirement, and the sensor and its specification are linked as design outputs. The supplier of the sensor has recently informed the medical device manufacturer that the sensor is reaching its end of life and will no longer be available in 6 months’ time. A change owner is assigned to identify and evaluate a new sensor. This person is most likely not the same engineer who originally designed and selected the first sensor. And that the original engineer may or may not still be with the organization, and may not remember why that sensor was selected. This is where having accessible and well managed requirements and risk management becomes important. The change owner can reference and look up the sensor, see the design inputs and risk with which it’s associated, and understand more quickly the criticality of the sensor and ensure the proper selection and testing are performed on a new sensor.
While change post-commercialization is inevitable, difficult change control management is not.
Management of requirements and risk extends through the entire life cycle of a medical device, including after a device has gained the necessary regulatory approvals and reached the market. Thus, take care in selecting the tools and developing the processes your organization uses for requirements and risk management.
In this post, we will discuss why start-up medical device companies should prioritize requirements and risk management before a quality management system.
As a medical device product development consultant, I often see start-up companies having trouble deciding what to prioritize – design controls and risk management or the quality management system (QMS). And what they mean specifically is, which software systems should the company invest in first – the requirements and risk management solution that will aid in building a regulatory compliant design history file, or the electronic-QMS system to establish the FDA required and ISO 13485-compliant QMS?
From my experience over the past 15 years, here are the 3 reasons why I advise start-ups to prioritize requirements and risk management over an eQMS system.
1. Design controls and risk management processes start earlier
For best product, schedule, and compliance success, incorporating design controls should be done proactively instead of reactively. Companies are typically developing their medical device from day 1. This is compared to other QMS processes that may not be used until years later when the product is being transferred to manufacturing and being commercially distributed. A few of these other processes include non‑conforming materials, device master record, product change control, and complaint management.
Thus, purchasing a full eQMS system earlier than necessary results in paying for functionality that may not be used for years. That is of low value to the start-up closely watching its funds.
In contrast, as the medical device is being developed from the onset of the company, the benefits of requirements and risk management solutions can be realized very quickly and much sooner than a full eQMS system.
2. Requirements and risk management is often unwieldy
Unless your device is ‘simple,’ for example no software, no electro-mechanical parts, low-risk Class 1 devices; thoughtful consideration should be given to the processes and solutions that will manage the various requirements and risk management for your medical device development. Organizing all the user needs, design inputs, regulatory requirements, requirements from industry standards, system requirements, sub-requirements, and risk management can quickly become unwieldy without proper management.
In my experience, even a Class II electro-mechanical device can easily approach a thousand line items to manage and connect. Add on embedded software or a digital interface, and that number can easily jump to multiple thousands of line items or more, depending on the complexity of the medical device. A solution like Jama Connect® has immediate value to ensure all items are linked, traced, verified, and validated for a regulatory complaint design history file and medical device file.
3. In the early years, a company can create and manage a regulatory compliant QMS without an all-electronic system
Does forgoing the eQMS mean settling with a non-compliant QMS? No. A company can implement a regulatory compliant QMS without an eQMS system. SOPs can be implemented in stages, prioritized on the stage of the company. These SOPs, along with a cloud-based document sharing repository, is often sufficient in those early product development years. As the company approaches transfer to manufacturing and commercial distribution, then is the time to evaluate whether it’s time to transition to an eQMS system.
Summary
In summary, these are the three reasons I advise start-ups to prioritize requirements and risk management first before an eQMS system. This path allows for the development of a successful product and complaint design history file, as well as establishing the rest of the quality management system, all in a practical manner that maximizes value and meets regulatory expectations.
In this post, we look at how Teledyne e2v leverages Jama Connect for improved communication and better risk management.
Teledyne e2v is a global leader in specialized components and subsystems for innovative solutions in medical, science, aerospace, defense, and industrial applications.
Over 1600 employees in countries across Europe, America, and Asia.
As a result of their track record of innovation and technological breakthroughs, Teledyne e2v continues to be involved in many high-profile ground-breaking programs
Teledyne e2v’s success is built on long-established relationships with industry partners. The combination of their strong, in-house technical capability and links with technical authorities and universities, ensures that they can bring together the right level of expertise for a diverse range of technical challenges.
They offer:
RF Power solutions for defense, medical, marine, industrial, and security applications
Imaging solutions including CCD and CMOS sensors and cameras, for space and earth observation, life science, machine vision, and medical applications
Semiconductor solutions for aerospace and defense hi-rel microprocessors, high speed data converters, and high reliability ICs
A Single Source of Truth Increases Product Development Efficiencies and Improves the Flow of Information for Teledyne e2v
To meet the challenge of producing components and systems with increasing complexity, Teledyne e2v turned to the easy-to-use Jama Connect platform to provide a single source of information and end-to-end traceability for some of their product development teams.
OBJECTIVES
Optimize information flow
Improve formal review process
Find a product development tool that would garner broad adoption
After selecting Jama Connect, Teledyne e2v saw these results
Time-Saving Efficiencies in Meeting and Decision Making
With the single source of truth and real-time structured collaboration that Jama Connect provides, the Teledyne e2v team has streamlined the development process resulting in increased cross-team efficiencies and expedited decision making.
Streamlining and Simplification of Tools
After determining that Jama Connect was a sensible, easy-to-use, and easy to adopt tool that would increase efficiencies, having a customer saying they were going to use Jama simply sealed the deal and is helping Teledyne e2v achieve their goal of streamlining and simplifying their tools and processes.
Jama Connect has enabled a focused model of engineering leadership, with identified owners of top-level vision, and a streamlined, single source of truth. This has shown strong potential to leave developers with greater freedom to manage the detailed development and implementation in-line with the vision that is clearly communicated and controlled within Jama Connect. Leveraging this further across all projects and product developments is a key ambition for the team.
Driving Systems Thinking Across the Organization
Teledyne e2v’s leadership knows that tackling complex engineering problems and projects requires a structured and systematic approach. They needed a tool that would support their engineers and technical specialists to work in a structured way—without the tool getting in the way. Jama Connect enables this and is helping to drive systems engineering discipline further into the team, not only standardizing the way people work, but making it a positive experience.
To see how the team is now leveraging Jama Connect and how their risk management process has already improved, read the full customer story here.
Editor’s Note: This post about how autonomous vehicles was originally published here on EngineerLive.com on October 6th, 2020, and was written by Jeremy Johnson, Jama Software’s Vice President of Product Management.
Never before have so many automotive engineers been tasked with bringing increasingly complex machines to market as they have with autonomous vehicles (AVs). Not to mention, it’s not simply a game of speed – elevating only the companies which manufacture products quickly – but a matter of those organizations taking these steps, while pushing the envelope on innovation and prioritizing consumer safety.
While important advancements are being made daily to bring fully autonomous vehicles into commercial availability, we are still a ways off from seeing the Level 5 autonomy we hope for. We’ve witnessed this in the significant challenges and shortcomings reported in the news in recent years, even amongst some of the biggest names in autonomous vehicle production. There are, however, a few actions engineers can take to stay nimble, innovative, and reduce the number of safety-critical mistakes throughout the development process.
Focus on your core business: advancing technology
This is particularly important for start-ups or companies looking to apply their technology to the automotive space for the first time. If you’ve heard the saying “don’t reinvent the wheel,” it comes into play when considering what processes and procedures for meeting industry best practices must be in place. Seek a consulting or technology partner that can enable your business to continuously practice requirements, risk, and test management in alignment with market standards such as ISO 26262 and ISO/PAS 21448. By starting with a proven framework that can be applied and moulded to a particular business, engineers can focus their innovation and organizational energy on delivering new technology to customers.
This focus is noticed in how Tesla drives its business, where the organization leans more heavily into internal development to drive technology advancement and differentiation in the marketplace. Audi, which attempted to meet Level 3 autonomy with its 2019 A8, sought outside suppliers such as Aptiv, Intel, Infineon and NVIDIA– a different approach to Tesla. Although Audi ultimately pulled back stating the car was too far into the lifecycle, it was realized through properly executed business and safety procedures.
Support collaboration internally and externally
Rapid innovation requires tight collaboration, often occurring across various hardware and software teams, and increasingly with partners or traditional competitors. Whether a formal joint venture or targeted collaboration around specific technology development, this “co-competition” has become more common as companies look to drive innovation in AVs forward.
Ensuring the tools and processes to enable this collaboration are in place, and capturing the critical output that comes with it, will guarantee that R&D efforts move quickly while maintaining strong focus on verification and validation. It’s especially important engineering teams get out of their silos and work with adversaries on this front, because in the U.S. in particular, there’s no mandatory compliance enforced by the government to follow standards such as ISO 26262 or SOTIF.
There have been promising signs of collaboration by some automotive companies to exchange learned information during AV development. One example is the collaboration among Aptiv, Audi, Baidu, BMW, Continental, Daimler, Fiat Chrysler Automobiles, Here, Infineon, Intel, and Volkswagen to develop a whitepaper, “Safety First for Automated Driving,” describing a potential framework for the development, testing, and validation of safe AVs.
Maintain traceability of requirements, tests, and risks
Developing complex, safety-critical systems that marry software and hardware requires a great deal of rigor and planning. Keeping track of each step of the development process with cumbersome documents and spreadsheets greatly hinders engineers’ ability to remain agile. By ensuring all of these steps are tightly managed, integrated into other product lifecycle phases and available for flexible reporting will enable organizations to innovate quickly. This also allows for prioritization of safety and compliance and the ability to rapidly adapt as the regulatory landscape continues to evolve..
Most countries do not yet have specific regulations that govern autonomous vehicles, leading to uncertainty around requirements, reporting, and future regulatory compliance. As previously mentioned, in the U.S. there is also an absence of tightly defined regulations which means states could implement differing standards that require specific nuances in technology and regulatory compliance. There’s also basic differences in infrastructure that makes development and safety a challenge – such as variability in road surfaces, lane markings, and signage.
The key for teams in this fluid environment is to remain close to standard development and regulatory agencies, as well as supply chain partners, to define and influence these regulations. And they must be prepared to show traceability of requirements, risk, and testing information in multiple formats to support the various potential points of oversight – be it downstream customers or regulatory auditors.
Ultimately, we’re likely a decade or more away from commercial availability of a Level 5 autonomous vehicle. As we inch closer, it’s vital engineering teams take advantage of the modern systems management tools at their disposal in order to get it right now – before it’s too late.
To see more information related to the automotive development industry, we’ve compiled a handy list of valuable resources for you!
Today we are excited to announce the availability of Jama Connect for Airborne Systems, a solution designed to help systems engineering teams reduce barriers to the compliance process for their aircraft and aviation systems development process.
Jama Connect for Airborne Systems lets customers seamlessly manage requirements, risks, and tests in one powerful platform while supporting mission and safety-critical standards. The solution is built with the digital engineering ecosystem in mind, empowering engineering teams to better manage requirements, while simplifying regulatory alignment for civil aircraft system development, including ARP4754/ED-79.
The aerospace and aviation industry is experiencing innovative changes not seen for decades, where rapidly evolving technology has driven companies to develop disruptive products. The first commercial greater than 50-seat hybrid-electric aircraft is expected to make a fare-paying flight by 20321 and the FAA estimates over 545,000 commercial drones to be in use by 2021.2Innovation brings increased complexity in the design process, including the connected networksthat handle autonomous flight systems and unmanned, autonomous aircraft. The industry currently relies heavily on paper documents to track requirements— which simply can’t be depended upon in a digitized world with predominantly autonomous aircraft.
The Chosen Solution for Leading Aircraft Companies
As the chosen requirements management platform for five of the leading global electric aircraft companies driving innovation, Jama Software recognizes and answers these challenges. The company has worked closely with its partners to provide an all-in-one solution to address and overcome these challenges with ease. Jama Connect for Airborne Systems helps engineering teams get set up quickly, allowing them to focus on product design and innovation, while reducing the costs and effort required to align their development processes to meet mission and safety-critical standards.
“Aerospace systems engineering teams have a tough job: they are tasked with developing innovative, mission-critical systems at an accelerated pace and with unwavering quality standards,” said Keith Johnson, Chief Solution Officer at Jama Software. “Our new solution, designed specifically for these teams, will help facilitate the development process from start to finish. Jama Connect allows developers to hit the ground running with a purpose-built, out-of-the-box framework and best–practices guides that save critical time in the engineering process.”
Key features of Jama Connect for Airborne Systems
Frameworks aligned with key industry standards and regulations: ARP4754/ED-79, DO-178C/ED-12C, and DO-254/ED-80 and SEBoK
Best practices, including procedure and configuration guides for aircraft and aviation systems development
Export templates and reports, including requirements specifications
Supply chain collaboration to enable an ongoing exchange of requirements between customers and suppliers
Training and documentation aligned to aircraft and aviation systems regulations, which provides accelerated onboarding to set systems engineers up quickly
To learn more about how Jama Connect for Airborne Systems helps teams to improve their ability to communicate, track, and test requirements for teams in the aerospace industry, download our solution overview.
With the rapid innovation in digital health such as clinical web and mobile software applications which do not traditionally classify as a “device” due to its lack of custom electromechanics, the Software as a Medical Device guidance (SaMD) was adopted by the FDA along with global regulatory leaders to ensure such software products adopt the same general medical device principles to demonstrate safety, effectivity and performance. The creation of SaMD was heavily influenced by new entrants unfamiliar with medical device regulations and terminology developing a broad spectrum of applications in order to support the new and growing industry.
On August 25th, 2020 Jama Software’s Clay Moore moderated a webinar featuring Beanstock Venture’s CEO, Shawnnah Monterrey, and past and present members of the FDA including John Murray and Bakul Patel.
Watch the full webinar now or read an abbreviated version of the transcript below.
My Software Is a Medical Device (SaMD). Now What?
Isabella Kennedy: As an industry, we are starting to see an influx of software applications being used for clinical support to inform clinical decisions, diagnosis, and treatment.We thought with all the new entrants unfamiliar with medical device regulations, it might be helpful to provide an overview and answer some questions. We’ve put together this amazing team of panelists that come from different organizations with tremendous offerings.
For example, BeanStock Ventures provides Software Online Agile Regulatory training, otherwise known SOAR, QMS, DHF audit and assessments, as well as medical device software development. Software CPR provides expert regulatory and standards compliance advice, FDA negotiations and strategies, and internationally recognized training courses.
We are all, of course, familiar with the FDA that provides numerous guidance documents related to software and specifically, in this case, SaMD. Jama provides software lifecycle management tools to aid in automating the process in facilitating compliance. As a follow-on, we will be sharing helpful resources from each company that might be available with you.
Let’s just have the panel members introduce themselves, starting with Shawnnah Monterrey.
Shawnnah Monterrey: Hi, I’m Shawnnah Monterrey, CEO of BeanStock Ventures.
Clay Moore: This entire category is around the definition and classification that drives regulatory status.
As technology evolves, and software becomes a platform and location independent, it is more important to understand what it does rather than where it resides. How does one determine if their software product is a medical device? Can you describe some elements of an intended use statement?
John Murray: Well, I think that everyone needs to understand that you can describe a device or a product in multiple, multiple ways. You can have engineering description, architectural description, all this stuff. But intended use is kind of special. It’s a special subset of all of that.
The focus of that description or that understanding, you need to focus on the patient and the practitioner. I think that’s the very important thing. I’ve seen lots of descriptions that don’t talk anything about the patient or the clinical use. When you’re addressing intended use, you may get confused by the definition and the regulation.
But you need to start asking yourself basic questions. Some of the questions I’ve come up with are, “So, I’m a doctor. How is this product going to make me or help me practice medicine better? How is this product going to help my patients live better lives?” Those focuses, clinical focused questions are very important to be honest about when you’re addressing that intended use statement.
That’s my first comment on that. Number two is that you have multiple customers here. You have the engineering staff, RAQA, marketing, and clinical staff. My suggestion before you tackle the intended use statement is to take a step back, and write a software product description, and get input from all four of those groups.
You need to balance the wheel and you’ll end up with a document that basically everyone can read, everyone understands. That will become the first step to risk management, the first step to design, the first step to regulatory status. It’s well worth the money and the time to do this correctly. The third caution I have about intended use is it’s never static. You start out today with an intended use and everything is well under control.
You design a great product, you have great customers, everything is fine, but before you know it, it’s being used in a different way. Your marketing team starts selling it in a different way. You need to make sure you keep that dynamic and alignment correctly. You don’t want to be in a situation where your intended use is stated this way, but your marketing intended use is a different way.
There are three things. One is to lean towards the clinical and patient. Write a full software product description. Number three; keep track of the dynamic because every SaMD that I’ve ever seen is always sliding into the future. It never stays the same because once you have it, you go, “I can use it for this now. This is a great opportunity so let’s move on.”
Clay Moore: That’s fantastic. I think we’ll spend some more time later talking about the iterative nature of SaMD products. Bakul, what’s some additional thoughts that you might have around intended uses?
Bakul Patel: Yeah, I think John hit up on most of it. I will just add to the concept of the clinical views and who the user is. From IMDR, and through work that we’ve done in defining and categorizing what Software as a Medical Device should look like and what’s a clear that definition looks like, I think we spend a lot of time talking about that.
Internationally, we thought about two things. What the software is doing. Where it’s going to be used. Who’s going to use it. How it’s going to be used. If you think about what the software is doing in the care, in the practice and then you have the other dimension of the context of sort of where it’s going to be used. Context is a bigger situation.
The term that we use in the IMDR framework is the healthcare situation and condition, which means it considers the disease, the condition of the disease through a varying spectrum of the disease you are trying to target. It’s not everything and anything. You can’t just say heart disease and cover the entire gamut because there are many things that you need to consider.
Even in there, there is a spectrum. Then you have the user who is going to use it in certain areas and certain types. Just working through the logic of where it’s going to be used, who’s going to use, and how it’s expected to be used. It’s sort of honing down on your intentions. I like the point John made about it’s not static. I’m going to give you a non-static version of what John was talking about.
Not just about static, about QMS. Start thinking you want to do this but then you find out your technology can only do this. There’s a difference between where you started and where you ended up. The result and what you want to put out to market should be exactly to what your product is performing. I think when that alignment happens between what the product does versus what you want the product to do is really what the regulatory terms calls intended use.
Clay Moore:Thanks for that additional context there. Shawnnah, what are some other thoughts you might have around intended use? Also, if you could start to peel back a little bit and talk about what if there are gray areas and we’re not sure about whether this really should be classified as SaMD?
Shawnnah Monterrey: Sure. I like to break down the intended use into four pieces. I like to focus on the user, the end user of the clinical product, the clinical utility of the product, the environment in which it’s going to be used, which could vary, and also the demographics of the patient.
Giving you an example, maybe I can walk through a simple example of maybe a heart monitor. A heart monitor, the environment would be, for this particular product, would be home use. The end user for that would actually be the patient. The demographic of the patient let’s say it’s age, I don’t know, 55 and up with heart condition that can be self-monitored.
The clinical utility is to monitor heart arrhythmia. Obviously, you would start there, and you can extrapolate further but I like to divide it in those areas to make sure all aspects of the intended use are covered. In terms of an example, I can give one particular example. I’m not sure if it’s still valid but if you consider Fitbit as traditionally not a medical device.
I think they’re moving towards getting it cleared or they may have cleared a version of it as a medical device in recent times. There was a lot of clinical applications being built on top of Fitbit. How do you determine if your product is a clinical device at that point if you’re taking data from the Fitbit and displaying it to an end user?
If you’re just taking data that’s being provided from the actual tool itself and you’re just passing it through, reading it basically, and rendering it, showing it on the display, generally, that’s not necessarily considered a clinical device. But if you take that data and you start providing additional analytics on top of it or start making clinical inferences based on that data, that’s the point where start getting into the medical device space.
Clay Moore: That’s great. What if the software helps the patient to not be in a clinical setting, if it’s used at home? John, maybe you’ve got some comments on the, again, would we think of this as something that’s being classified as SaMD or not?
John Murray: I don’t think that the device, not a device question starts with at home use or in clinic use. I think that’s a secondary question. It may determine the actual classification of the product. The product may be for monitoring blood glucose, take an example. You could have one product that’s for design to be used in a clinic, one for use at home. They may have different risk determinations.
That may make the classification different. It always must start with the specific intended clinical use, in my opinion. Part B would be now what’s the risk of this application? That varies depending upon the patient population, the environment of use, and a bunch of other questions that could come into being. That would be how I would start framing that up as a question.
Clay Moore:That’s great. Bakul, any other commentary on example of things that are SaMD or aren’t SaMD? Maybe some gray areas.
Bakul Patel: Yeah. Recently, in the act or jurisdiction doesn’t extend to things that are not meant for clinical purpose. It must be for caring, mitigation, treatment or preventing a disease. That’s statutory definition.
An activity tracker just measuring how many steps you’re taking per day and helping you stay healthy is not something we would consider as a medical device even from that perspective. Then certainly, I think this is what Shawnnah was talking about, is you take that steps and say because you took X, or you took less or took more steps, it means certain disease, or it means certain condition.
Clay Moore: Great. I think each of you mentioned risk at some point in time during each of your responses. Let’s move into more the things that are related to risk and the amount of effort that’s behind these things. Once a company determines their software product is a medical device, the next step is to determine its risk category.
Risk categories are used to determine the rigor required to ensure that the product is safe, is effective, and it needs to perform its criteria necessary. For SaMD, risk is classified using a framework where one is the lowest risk and four is the highest risk. John, can you discuss why understanding risks is important when developing and maintaining a software to medical device?
John Murray: Well, the fundamental basic idea is you don’t want anything bad to happen to good people. That’s the purpose of all these rules. We want to make sure your product doesn’t hurt anybody in a way that’s preventable. That’s the simple explanation of that. I want to talk a little bit about one of the special elements of SaMD in my opinion.
That is that we have a specific risk category that I often see with many of my clients gets left off. I’m going to call that engineering or environmental risk, which is a little bit different than clinical risk, traditional clinical risk. Because you know you’re using a SaMD, you need to think about all the elements of that platform choice that may have an impact on harming people.
It may have an impact on risk. The beauty of this is that once you set up this model, you can reuse these questions repeatedly with every product. One simple example is what if the Bluetooth connection becomes lost? What kind of impact is that going to have on my patient? What kind of impact is that going to have on my delivery of my intended use?
All the known features of a general-purpose computing platform, wi-fi, Bluetooth, all that kind of stuff, you can ask those questions. The beauty of this is that I know the engineers are asking these questions all the time. Those questions never get translated over to the regulatory side where the risk management thing is set up.
The idea is that you want to do what you can do. Maybe if you lose Bluetooth for five seconds, it’s not a big deal but what if you lose it for an hour or two hours? There’s a huge number of SaMD applications where you can lose that connection for a short period of time. It’s not going to be an impact. Do you need to know when that happens, and do you need to inform the patient when that happens? In some applications, a patient needs to know they’re not connected.
One of the other ones that I thought of and we saw in some recalls was that what if you start your algorithm, and you are resource deprived? You don’t have the right amount of memory.
You don’t have the right amount of algorithm power, computing power. All these questions, I know they get asked all the time by the engineering staff. The engineering staff needs to be accustomed to contributing that information to the design and to the regulatory affairs side so that when you present your case, you’ll say, “Yeah, we covered all the environmental engineering risk and here’s our list here.”
That gets sandwiched together with the clinical risk, which I’m not a doctor so I don’t like to talk about that very much. That’s a hugely important thing, is being honest. Being honest that bad things can happen, but will they have an impact on my clinical application?
Clay Moore:Shawnnah, why don’t we go over to you and would love to get some additional color on that as well as maybe thinking about what are some aspects that influence safety, effectivity, or performance?
Shawnnah Monterrey: When I look at performance, I think about technological performance and analytical performance. From a technical perspective, especially for a SaMD product where it can be hosted in numerous environments, you really want to go back and understand how the operator is going to be using your system and the impact that that environment may have on your product.
I’ll give you an example of something that could impact performance for a hosted cloud application could be the latency of the network. Something you can’t control, so it’s important to really understand the environment in which your product is going to be used and then how that would then influence some of the safety associated with some hazards to be identified.
A top four list can include stuff like no result. What would happen if the network goes down and the end user wasn’t able to receive results? Delayed results as a result of network latency and is there any impact to false positive or false negative as a result? I think network latency may not directly influence a false positive and false negative butresults in a no result or a delayed result.
That’s something that you should assess and determine the severity based on, again, the intended use of your product. From another performance perspective would be the analytical side. That’s focused more on the sensitivity and specificity of your product and really, looking at a false positive and false negatives.
That requires, again, an understanding of your demographics. What is that patient population that is needed to be able to truly test your product to show and prove the analytical performance of the system?
Clay Moore:There was a question around the differences and overlap between SaMD when it’s a mobile medical application and software is inside a medical device considering the same software can be leveraged across different user contexts.
Bakul Patel: I think the intended use of a piece of software does not rely specifically on the platform that it’s residing on. I think that’s one thing you have be very clear about.
The software is going to take advantage of the computing resources and the surroundings of that environment is what I call it. I don’t think it matters from that perspective. Whenever somebody makes a choice to put a product out, they intentionally market it or design it for a particular product or platform.
When you choose that, I think you need to make sure that you know all the things that you are going to depend on or not depend on in that platform. It could be a multi-thread application or a multi-thread operating environment, but you may not use multi-thread for all kinds of various reasons.
You may know using multi-threading applications may lead to some issue that may raise conditions and you may want to think about that. When you start using operating systems and you are using instructors and you have multi-course going on today, you are going to have to say that, “Even if I used a simple programming language to be on a certain platform, the platform may use my program separately because you have different threads running.”
Just knowing that will help you start answering some of the questions about what do you have to mitigate? How to reduce the risk to a minimum. How do you consider that? I think cloud services is very similar. I think you have very different challenges. Mobile is the same. Very different challenges and different sort of advantages. You just take those into account.
To see more information specific to the medical device development industry, we’ve compiled a handy list of valuable resources for you!
![[Webinar Recap] Lessons Learned for Reducing Risk in Product Development](https://www.jamasoftware.com/media/2022/02/2022-02-17-reduce-risk-product-development.png "[Webinar Recap] Lessons Learned for Reducing Risk in Product Development")