In this blog, we offer a preview of our comprehensive guide to understanding ANSI/AAMI SW96:2023 and mitigating security risks.
What You Need to Know: ANSI/AAMI SW96:2023 — Medical Device Security
Managing risk around a medical device’s entire lifecycle has become increasingly complex. Many devices use third-party components, which is especially true for devices that require a network to operate. This increased need for connectivity, along with other emerging threats, is putting security at the forefront of medical device industry standards.
A recent report titled “2023 State of Cybersecurity for Medical Devices and Healthcare Systems” found 993 vulnerabilities in the 966 medical products it examined—a 59% year-over-year increase from 2022. Software applications, including those that medical devices relied on to work, accounted for 64% of the vulnerabilities found.
With device vulnerability increasing, new standards aim to keep up with emerging threats. As a result, ANSI/AAMI SW96:2023 was created to help protect against threats, understand risk, and guide manufacturers in taking the most appropriate actions to enhance security. However, because the standard is relatively new, many device manufacturers are still finalizing the interpretation of how this impacts their organizational processes. If you’re still working to get familiar with the standard, we’ve created a complete guide to make the task easier.
THIRD-PARTY COMPONENTS MAY INCREASE SECURITY RISK, WITH ONE STUDY FINDING THAT SOFTWARE ALONE ACCOUNTED FOR 64% OF NOTED VULNERABILITIES.

What is ANSI/AAMI SW96:2023?
ANSI/AAMI SW96:2023 guides security risk management for medical devices, aligning with the processes included in ISO 14971:2019.
The new standard addresses the entire lifecycle of a medical device, including areas such as design, production, and post-production. It’s intended for use with AAMI TIR57 Principles for Medical Device Security – Risk Management, which addresses cybersecurity analysis, and AAMI TIR97, Principles for Medical Device Security, which guides processes for managing medical devices in the post-market space.
The goal of the new standard is to support manufacturers in ensuring that medical devices are reliable, work as intended, and don’t cause harm to patients, operators, or the environment. It also focuses on mitigating any potential risks around device failure.
RELATED: Understanding Integrated Risk Management for Medical Devices
Why is security for medical devices important?
Security has always been important to medical device manufacturers, which is why considerations are included in ISO 14971:2019. However, ANSI/AAMI SW96:2023 aims to deepen security-related standards.
Addressing potential security risks throughout the entire product lifecycle, including design, production, and post-production, enables manufacturers to identify and mitigate potential risks through a more focused and proactive approach. It helps manufacturers continually identify, review, and safeguard against fast-evolving threats
Understanding the security risk management process
As you get up to speed with ANSI/AAMI SW96:2023, the “security risk management process” section includes details for mitigating potential threats. It includes six major sections, everything from security risk analysis to production and post-production activities. Each section contains a detailed framework, but for the sake of simplicity, we’ve highlighted a few main points for each.
The 6 Sections of Security Risk Management
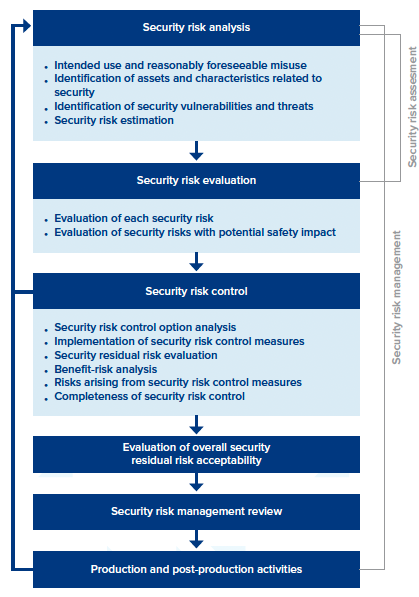
- Security risk analysis. It focuses on selecting product security standards, performing threat modeling, and establishing capabilities to identify and detect security vulnerabilities across a medical device’s entire lifecycle.
- Security risk evaluation. Establishes a security assessment strategy and testing processes.
- Security risk control. Identifies, designs, and implements security risk control measures, as well as verifying the implementation effectiveness of any security risk control measures.
- Evaluation of overall security residual risk acceptability. Determine if the “security residual risk” of a device is acceptable.
- Security risk management review. A security management report is prepared.
- Production and post-production activities. Potential vulnerabilities are monitored to identify any new security risks. Also, it establishes processes to stay aware of new threats, creating security incident response plans and other measures to identify ongoing vulnerabilities.
Section 1: Security Risk Analysis
The security risk analysis focuses on selecting product security standards, performing threat modeling, and establishing capabilities to identify and detect security vulnerabilities across a medical device’s entire lifecycle. It covers:
- Security risk analysis process: It suggests that manufacturers perform a security risk analysis, and the results are recorded in the “security risk management file.”
- Intended use and reasonably foreseeable misuse: The “security risk management” file includes reference documents developed in compliance with clause 5.2 of ISO 14971. It needs to account for “the use of a medical device in a way not intended by the manufacturer, but which can result from readily predictable behavior.”
- Identification of assets and characteristics related to security: You’ll also identify potential medical device vulnerabilities such as third-party components, hardware, and software.
- Security risk estimation: You will estimate the associated “risks” for each of the identified security vulnerabilities and potential impacts on areas like confidentiality and integrity.
Section 2: Security Risk Evaluation
The security risk evaluation establishes a security assessment strategy and testing processes. A few areas it considers:
- Evaluation of each security risk: Identify each security risk area, determining if a “security reduction” is required.
- Evaluation of security risks with a potential safety impact: Consider every potential risk to determine any potential safety impacts.
RELATED: The Complete Guide to ISO 13485 for Medical Devices
Section 3: Security Risk Control
This section is focused on identifying, designing, and implementing security risk control measures, as well as verifying the implementation effectiveness of any security risk control measures, including:
- Security risk control option analysis: Determine if a security risk control measure is appropriate for mitigating security risks to an “acceptable level.”
- Implementation of security risk control measures: Security risk measures are selected based on the prior step.
- Security residual risk evaluation: After the security risk control measures are implemented, the manufacturer evaluates the security residential risk and records this evaluation in the security risk management file.
- Benefit-risk analysis: If a security residual risk is found to be “acceptable” using the criteria created in the security risk management plan, and further security risk control isn’t practical, the manufacturer conducts benefits versus security risk analysis.
- Risks arising from security risk control measures: The manufacturer reviews the effects of the security risk control measures to understand whether new security vulnerabilities and threats are introduced that could impact security, safety, or privacy.
- Completeness of security risk controls: The manufacturer periodically reviews security risk control activities to ensure all vulnerabilities and threats are considered and security risk control activities are complete.
Section 4: Evaluation of Overall Security Residual Risk Acceptability
After the security risk controls are implemented and verified, the manufacturer determines if the overall “security residual risk” created by the medical device is acceptable.
Section 5: Security Risk Management Review
The standard recommends a review of the execution of the security management plan before releasing a new device. According to ANSI/AAMI SW96:2023, the review should ensure:
- The security risk management plan has been appropriately implemented.
- The “security residual risk” is at an acceptable level.
- Methods are in place to gather and review details in the production and post-production phases, and leadership has reviewed and approved the plan.
Section 6: Production and Post-production Activities
The final section is focused on establishing, documenting, and maintaining a system to monitor, assemble, and review information about medical device security in the production and post-market phases. Also, it establishes processes to stay aware of new threats, creating security incident response plans and other measures to identify ongoing vulnerabilities.




![[Webinar Recap] Leveraging Jira & Jama Connect® for Enhanced Requirements Management](https://www.jamasoftware.com/media/2024/04/Beyond-Jira-Word-Elevating-Requirements-Management-1.png "[Webinar Recap] Leveraging Jira & Jama Connect® for Enhanced Requirements Management")



![[Webinar Recap] Concurrent Engineering in Aerospace and Live Traceability™](https://www.jamasoftware.com/media/2024/04/Concurrent-Engineering-in-Aerospace-and-Live-Traceability™-1.png "[Webinar Recap] Concurrent Engineering in Aerospace and Live Traceability™")