This blog overviews our Datasheet, “Simplify Complexity, Risk Assessment, and Safety and Cybersecurity Compliance with Jama Connect for Industrial Machinery Development”
KEY BENEFITS
- Streamline Standards Compliance: Automate the traceability required for standards, significantly reducing the manual effort of audit preparation.
- Support Secure-by-Design: Seamlessly incorporate cybersecurity planning and controls from design initiation to ensure compliance with EU Cyber Resilience Act requirements.
- Adopt Agile Approach to Contextualize Functional Safety Assessments: Customize assessments to fit each specific product or iteration instead of using the same preset list of hazards and responses for every project.
- Unify Risk Management: Integrate hazard analysis (HARA) and Failure Mode and Effects Analysis (FMEA) directly into the development process to ensure safety risks are identified and mitigated early.
- Enhance Multi-Disciplinary Collaboration: Align mechanical, electrical, and software teams on a single platform to prevent silos and ensure system-wide coherence.
- Accelerate Variant Management: Manage product variants efficiently to meet specific customer specifications without sacrificing speed to market.
- Ensure End-to-End Traceability: Maintain links between requirements, risks, and tests to ensure every design decision is verified and validated before release.
Simplify Complexity, Risk Assessment, and Safety and Cybersecurity Compliance with Jama Connect for Industrial Machinery Development
Developing modern industrial machinery involves navigating a dense web of complexity where precision is paramount. Engineering teams must synchronize mechanical, electrical, control, and software components while adhering to rigorous safety and security standards like ISO 13849-1 and 2, IEC 62061, IEC 61508, and IEC 62443. The pressure to deliver tailored product variants rapidly often conflicts with the need for thorough risk assessment and documentation. Without a unified approach, gaps in requirements can lead to costly delays, safety incidents, or field recalls, threatening both market reputation and operational efficiency.
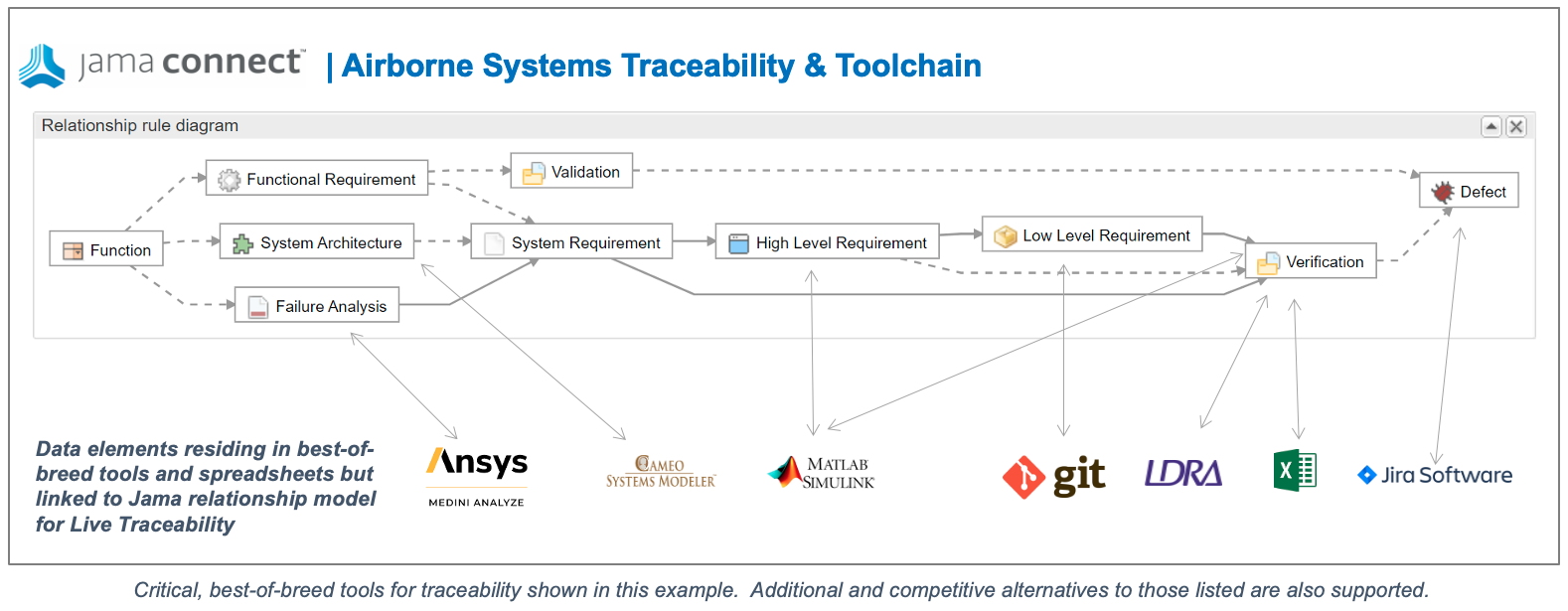
Jama Connect for Industrial Machinery Development provides a robust, pre-configured framework designed to tame this complexity. By aligning directly with major machinery and functional safety and security standards, the platform creates a clear digital thread from high-level stakeholder requirements down to specific component verification. This solution bridges the gap between diverse engineering disciplines, ensuring that control systems, safety functions, and mechanical designs evolve in lockstep. Teams manage the entire product lifecycle — from concept to validation — within a single source of truth that actively monitors for compliance and risk.
RELATED: Agile Robots Boosts Internal Process Efficiency by Moving to Jama Connect
Jama Connect for Industrial Machinery Development includes the following:
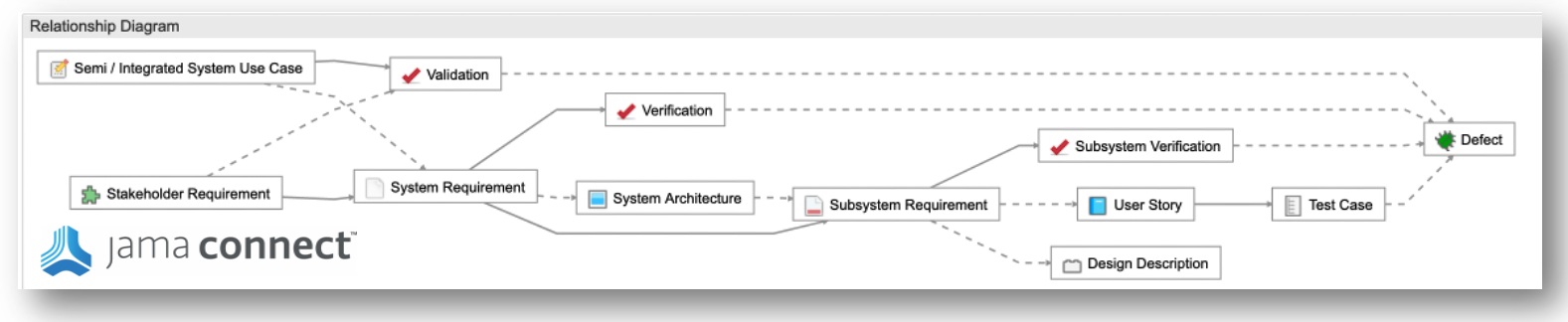
- End-to-End Traceability. The out-of-the-box, customizable Traceability Information Model™ starts right at the top with every stakeholder or customer requirement tracing back to a specific standard or clause. This traceability provides teams with a clear link between what they’re building and why it’s required, and detailed documentation for auditors.
- Functional Safety Compliance. The classic V-model structure covers stakeholder to system, subsystem, component, design, and then test for a clean, end-to-end chain that mirrors the safety lifecycle — define it at the top, prove it at the bottom.
- Integrated Cybersecurity Framework. Identify relevant threats and vulnerabilities using pre-defined templates to align threat analysis with security requirements and verifications, enabling teams to respond to incidents quickly at all stages of the product lifecycle.
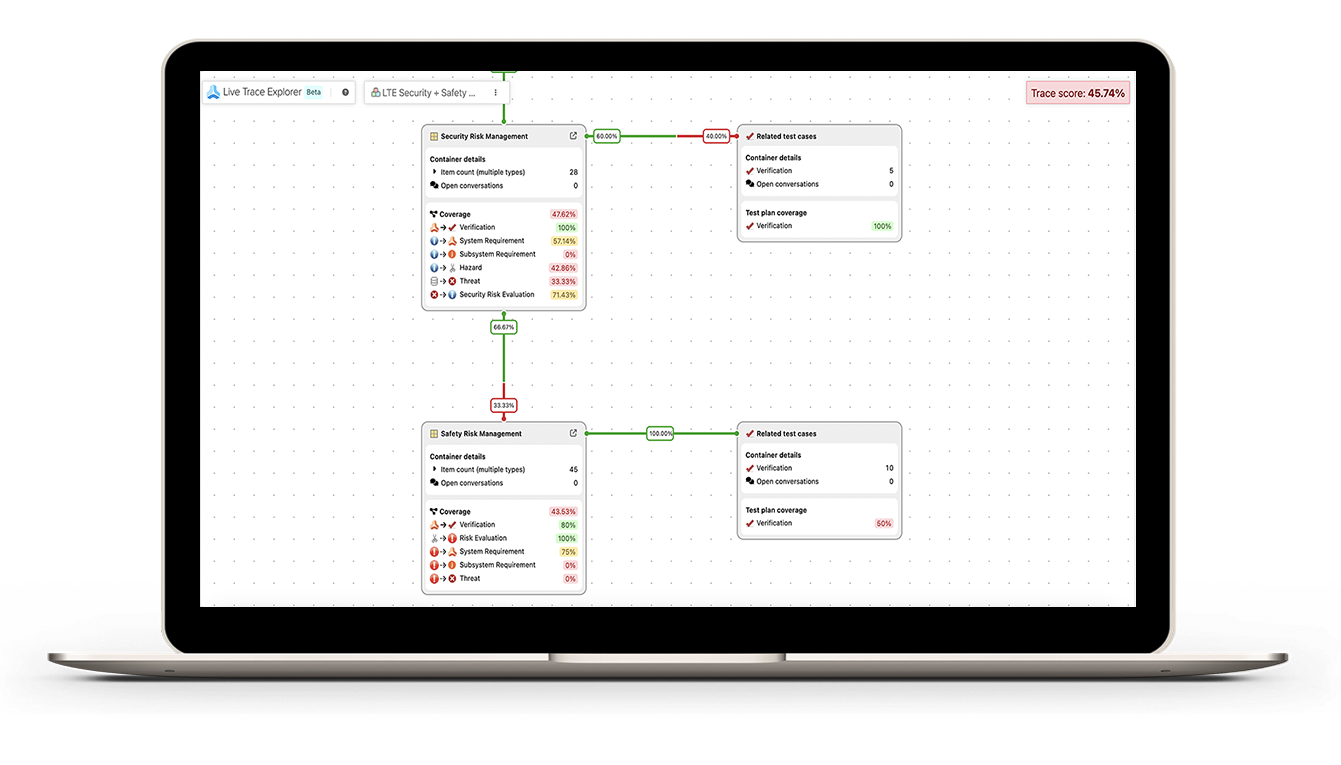
- Risk Management. Each use case connects into a hazard analysis or FMEA, which flows naturally into safety function requirements. That means that identified risks turn directly into design actions, not just documents that sit on the shelf.
- Control Systems Safety. Safety functions break down into the safety-related parts of the control system — electrical, electronic, or software layers, where things like Performance Level or SIL come into play.
- Verification and Validation. Every safety function, every requirement, has a clear link to the tests or activities that prove it’s been met.
From standards, threats, and risks all the way through design and verification, everything is connected. It makes compliance smoother, audits faster, and the overall process a lot more reliable and efficient.
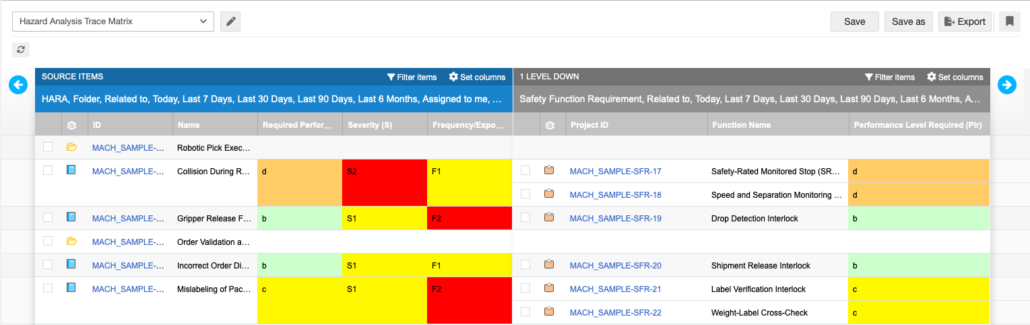
Example of Hazard Analysis Trace Matrix
Companies choose Jama Connect for Industrial Machinery Development to innovate faster and deliver complex, safety-critical machinery with confidence, knowing that every requirement is met, tested, and documented for the global market. To learn more, visit www.jamasoftware.com