Design history files were first required by the FDA in 1990 for medical devices. Learn how DHFs impact product teams, and get Jama Software’s DHF template.
In the competitive world of medical device development, medical device companies can’t afford to lag behind the pack. Streamlining the design and approval process is vital to getting products to market in a timely manner. Proper documentation is key to a smooth approval process.
One essential ingredient to medical device documentation is the Design History File, or DHF. While a bad DHF may not result in non-compliance, a good DHF can make the difference between a smooth compliance process and a bumpy one. To create a good DHF that streamlines compliance, design teams should know what to include—and what to avoid—as they go through the entire design and approval process.
What’s a Design History File, and How are They Used by Product Teams Working on Medical Devices?
Before reviewing all of the essential DHF ingredients, it can help to back up and establish what it is and why it’s important.
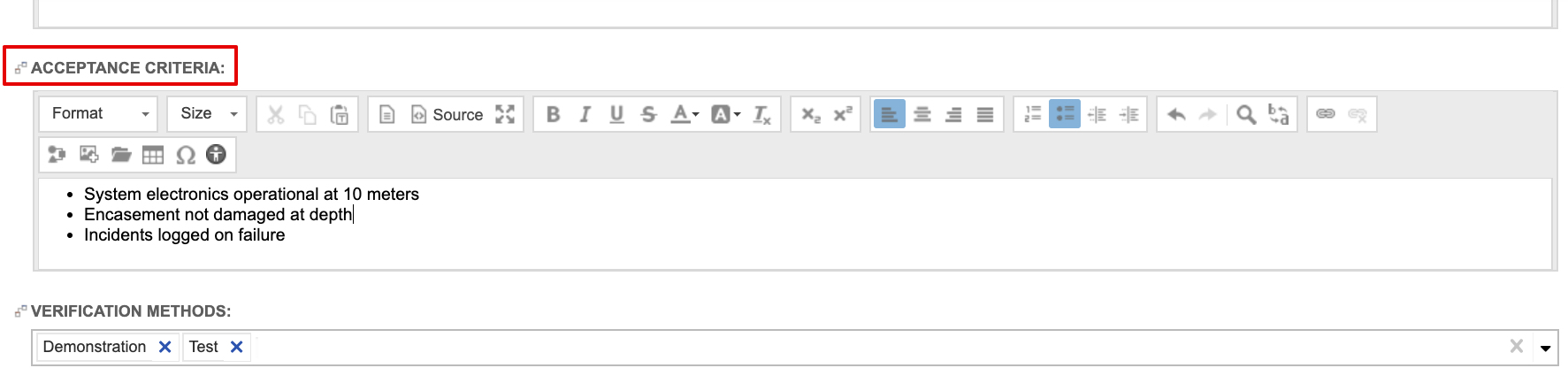
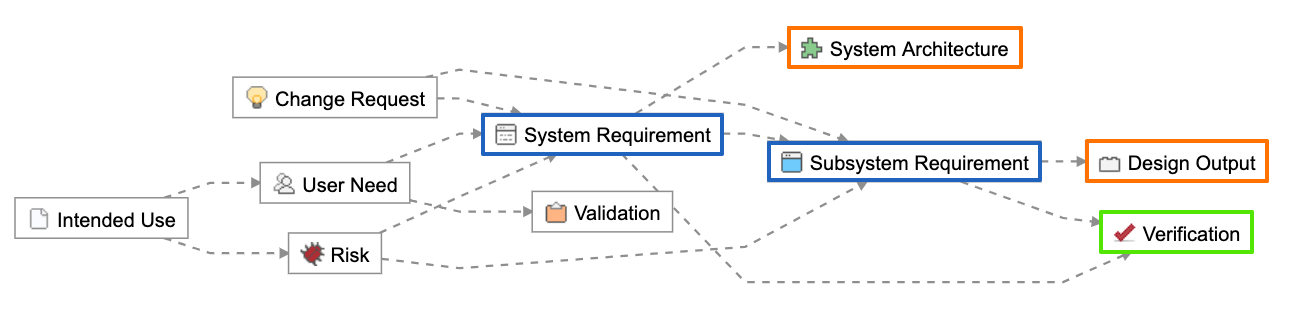
The Design History File is a collection of documents that describe the design and development activities of a medical device. Its purpose is to demonstrate that the device was developed using the design control process needed to meet FDA requirements.
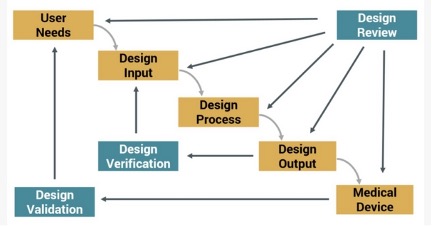
Figure 1: Design Control Process for Meeting FDA Requirements
The DHF requirement was established as part of the Safe Medical Devices Act of 1990. The act describes the DHF in section 21 CFR 820.30, sub-section (j) as follows:
“Each manufacturer shall establish and maintain a DHF for each type of medical device. The DHF shall contain or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plan and the regulations of this part.”
There are two key terms in the DHF requirement: “establish” and “maintain.” “Establish” means that the development team must define, document, and implement requirements. Evidence is found in procedures, work instructions, drawings, and other similar documents. “Maintain” means that the team must review, approve, and update requirements. Evidence of maintenance is typically found in document control or change control systems of a company.
While a comprehensive DHF is vital to the FDA approval process, design teams will realize other benefits of maintaining the DHF, including:
- Requirements traceability;
- Change control; and
- Knowledge preservation and information sharing among key stakeholders.
What is the difference between DHF and DMR?
The DHF is one of three pillars of design compliance for medical devices:
- Design controls: The moving parts of product design and development.
- Design History File (DHF): The collection of records from design and development activities.
- Device Master Record: The “recipe” for product manufacturing once the design is complete.
As one of the pillars of design compliance, the DHF is important, but it cannot hold up the design process on its own. It’s one leg of a tripod, and removing any one of the legs means that the entire design compliance process may tip over. Equally important to the process are design controls and the Device Master Record, or DMR.
What are design controls, and how does the DHF establish the design control process?
Design controls establish a plan to manage new product development; they are the process pieces that guide development along a predictable path to ensure that requirements are met. The Safe Medical Devices Act requires that manufacturers of Class II and Class III medical devices (and some Class I devices as well) implement design controls over the development process.
Design controls include:
- Design inputs: The physical and performance requirements for the type of device. May also include types of materials needed, interfaces with other devices, and even packaging required, especially if the device must be sterilized.
- Design outputs: Usually the final specifications for the device, typically documented in models, drawings, engineering analysis, etc. Outputs should trace directly back to initial requirements. Design outputs are the basis of the Design Medical Record (DMR).
- Design review: A formal review by the design team and any other relevant parties, such as sales, marketing, manufacturing, etc. Documentation of the review should include the date, participants, version/revision reviewed, and results.
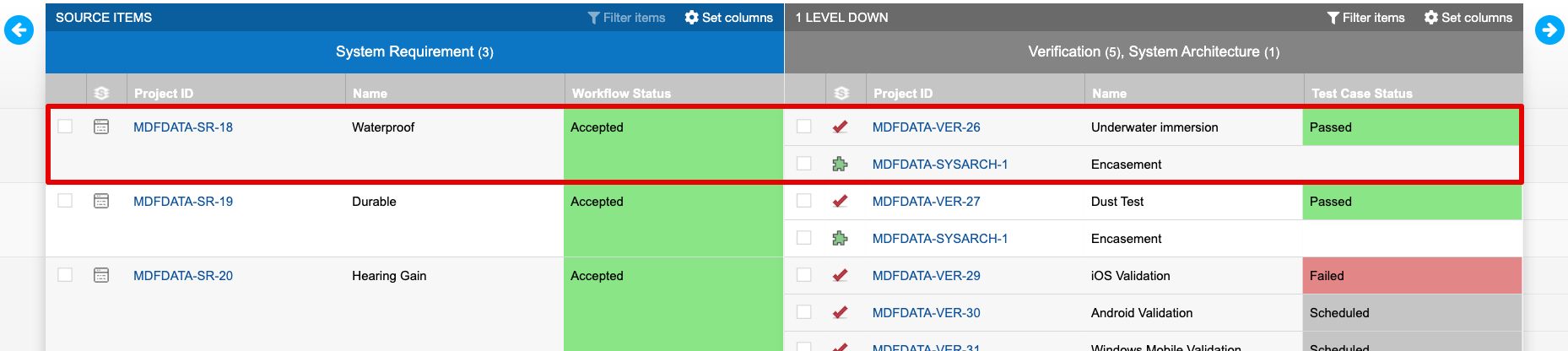
- Design verification: This process verifies that the design output meets the requirements expressed by the design inputs and that specifications are correct. Documentation of this process must include the date, participants, version/revision reviewed, and the verification method and results.
- Process validation: Process validation involves producing the device with a normal, low-level production process to confirm that the device will function as designed outside of the prototype process. Documentation of the process is required for the DHF.
- Design validation: In this process, each manufacturer validates device design under specific conditions on small production lots or batches. The DHF documentation must include identification of the design, methods of production, date, and individuals performing the validation.
- Design transfer: The process of transferring design into production, distribution, and installation specifications.
- Design changes: May be referred to as “engineering change” or “enterprise change,” this process identifies and documents any design changes.
- Design history file: This is the formal document prepared for each medical device that includes either all documents generated in product development process OR an index of documents and locations where they are stored.
As you can see, the DHF is one of many design controls, but it also serves as the repository of records of all other design controls. Think of it as the final stop for the information generated by all other design controls.
What is in a Device Medical Record (DMR)?
The third leg in the tripod supporting your compliance process is the Device Medical Record, or DMR. Think of the DMR as the “recipe” that resulted from the design controls to manufacture the device according to design specifications. It’s more than just a production traveler—it will include device specifications, compositions, drawings, formulations, software specifications, etc., as well as production process specifications, including appropriate equipment, methods, procedures and environments, such as clean rooms, humidity controls, quality assurance procedures, and any specialized equipment needed. The DMR will include everything from beginning to end of the production and shipping process, including any relevant installation instructions.
What are some pitfalls to avoid when compiling the DHF?
Many companies make costly and time-consuming errors when creating their DHFs, leading to longer-than-necessary audits and failed FDA inspections. Avoid these mistakes, and you’ll minimize lost time and unnecessary costs getting to market.
Jama Software hosted a webinar led by Quality Centric Consulting to review:
- The top DHF errors that are costing you money
- How to tackle 21 CFR 820.30 requirements
- How Jama Connect™ streamlines the DHF process

Five of the most common pitfalls of the DHF include:
Not having a DHF: Usually small companies strapped for resources will put all their efforts into designing and getting a product to market, with the intention of doing the DHF later on. This could be due to a lack of understanding of what DHF outputs are required.
Chaotic and unorganized DHF: This is more typical in paper-based companies that keep all information in binders, but leave off dates or include unapproved documents, making it impossible to organize at the time of an audit or inspection. Disorganization — whether paper- or email-based — also makes it difficult to manage changes and revisions due to information silos and lack of sharing information.
Including unnecessary documents: Adding business documents, such as supplier quotes, financial information or unused concepts, can bring a flurry of unwanted questions from inspectors. This can also be a symptom of not having a DHF tool or template, as well as a linear process that involves lengthy review and approval times.
Unmaintained DHF: Often companies use either outdated or improper technology for the job. Paper-based systems, email, shared folders with read-write access and the like, are not always effective document controls. This can lead to lost copies of important records, making a DHF non-compliant.
Not traceable to outputs: Without a DHF, it’s impossible to create your DMR, and very difficult to establish your traceability matrix. If design control results are not well documented, easy to locate or current, your DMR will fail to be compliant.
RELATED POST: Avoid the Most Common Challenges of the Design History File
Creating the DHF may feel like a heavy burden, but it is the key to a smooth compliance process. By treating the DHF as part of your design and development process from the beginning, these requirements become seamless and help ease the path to compliance.
Jama Connect can help traceability and requirements management and encourage a systems approach to medical device development, making the creation of a DHF more seamless and streamlined than ever before. To learn more, contact Jama Software.
To learn more about how Jama Software can help Product Teams stay compliant, visit Jama Connect Solutions for Medical Development