
In a recent webinar about the common pitfalls of the Design History File (DHF), experts discussed what medical device manufacturers need to know about design compliance. Below are some highlights.
Three Pillars of Design Compliance
There are three pillars of design compliance for medical devices:
- Design Controls: The moving parts of product design and development
- DHF: Collection of records from design and development activities
- Device Master Record (DMR): The “recipe” for product manufacturing once design is complete
In order to achieve FDA compliance and effectively bring your product to market, you must successfully satisfy each of the three pillars.
What is a DHF?
The DHF is a compilation of documents that describe the design and development activities of a medical device. Its purpose is to show that the medical device was developed using the design control process, pictured below, needed to meet FDA requirements.
Design Control Process for Meeting FDA Requirements
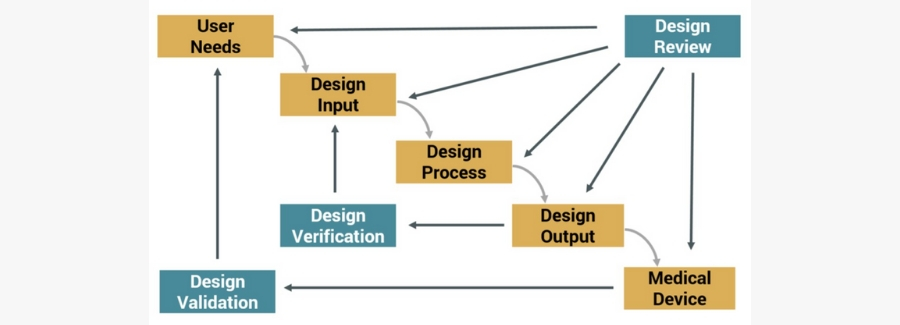
The FDA lists specific guidelines for DHF under 21 CFR 820.30 (j):
“Each manufacturer shall establish and maintain a DHF for each type of medical device. The DHF shall contain or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plan and the regulations of this part.”
The DHF also provides traceability to requirements, can assist with change control and acts as a tool for knowledge preservation and information sharing within your organization or with your supply chain.
Complications of an Incomplete DHF
As the second pillar of design compliance, the DHF connects the first and the third pillars. As noted, it documents that the correct design control process was followed. The DHF is also the basis of the DMR, which must be created according to design specifications.
An incomplete DHF makes it nearly impossible to prove you used the correct design control process for meeting FDA compliance, or create a compliant DMR.
Furthermore, if there are deficiencies noted within the design control subsystem during an inspection, they are documented by the FDA. If your company’s response is inadequate or not timely – which is likely when your DHF is lacking – a public warning letter could be issued. This could not only hurt your credibility in the market, but it might also give your competition some insights that you’d rather have kept within your company’s walls.
Five Common Pitfalls of the DHF
According to experts, these are some other reasons a DHF might fail:
Not having a DHF
Usually small companies strapped for resources will put all their efforts into designing and getting a product to market, with the intention of doing the DHF later on. This could be due to a lack of understanding of what DHF outputs are required.
Chaotic and unorganized DHF
This is more typical in paper-based companies that keep all information in binders, but leave off dates or include unapproved documents, making it impossible to organize at the time of an audit or inspection.
Disorganization — whether paper- or email-based — also makes it difficult to manage changes and revisions due to information silos and lack of sharing information.
Including unnecessary documents
Adding business documents, such as supplier quotes, financial information or unused concepts, can bring a flurry of unwanted questions from inspectors. This can also be a symptom of not having a DHF tool or template, as well as a linear process that involves lengthy review and approval times.
Unmaintained DHF
Often companies use either outdated or improper technology for the job. Paper-based systems, email, shared folders with read-write access and the like, are not always effective document controls. This can lead to lost copies of important records, making a DHF non-compliant.
Not traceable to outputs
Without a DHF, it’s impossible to create your DMR, and very difficult to establish your traceability matrix. If design control results are not well documented, easy to locate or current, your DMR will fail to be compliant.
If it feels like creating a DHF is a heavy burden, you should seriously consider the alternatives as well as the urge to make it an afterthought. If you treat your DHF as part of your design and development process, these requirements become seamless and help ease the path to compliance.
To learn more about preventing these issues in your DHF, watch the webinar, Common Pitfalls of the Design History File and How to Avoid Them.
Subject matter experts from Jama Software provide guidance and best practices on requirements management, test management, compliance and regulation, risk management, and complex product and software development.
Latest posts by Jama Software (see all)
- G2® Once Again Names Jama Connect® the Overall Leader for Requirements Management Software for Spring 2024 - April 18, 2024
- FDA Outlines its Approach to Artificial Intelligence Regulation - April 16, 2024
- Manage by Exception: Data-Driven Practices to Improve Product, Systems, and Software Quality - April 11, 2024