Jama Software is always on the lookout for news and content to benefit and inform our industry partners. As such, we’ve curated a series of articles that we found insightful. In this blog post, we share content sourced from HealthITSecurity – MDIC, HSCC Team Up to Establish Medical Device Security Benchmarks – which was originally published on June 2, 2022, by Jill McKeon.
MDIC, HSCC Team Up to Establish Medical Device Security Benchmarks
Experts from MDIC, HSCC, and BD discuss a new self-assessment tool that aims to establish medical device security benchmarks.
The Medical Device Innovation Consortium (MDIC) and the Public Health Sector Coordinating Council (HSCC), in partnership with Booz Allen Hamilton, created a new survey with the goal of establishing medical device security benchmarks.
Medical device security continues to be a pain point for healthcare organizations, regulators, and manufacturers. The sheer number of devices on an organization’s network at any given time, along with the prevalence of legacy devices and a lack of industry-wide standards, have posed significant security challenges.
Over the years, there has been a lot of finger-pointing and confusion surrounding roles and responsibilities for medical device security.
“There was no mutual understanding about shared responsibility between device manufacturers, hospital systems, and healthcare providers,” Greg Garcia, executive director for cybersecurity at HSCC, explained in an interview with HealthITSecurity.
“We quickly recognized that as a sector, we needed to be doing something about this rather than just staying in our corners.”
In an effort to address these concerns and promote shared responsibility for medical device security across the industry, the HSCC Joint Cybersecurity Working Group (JCWG) issued the Joint Security Plan (JSP) in 2019. The JSP is essentially a product lifecycle reference guide to developing, deploying, and supporting secure medical devices and health IT products and solutions.
“The JSP is expected to evolve over time and the HSCC intends to establish a governance model to ensure the baseline strategy is updated based on execution of existing plans or new needs identified by members of the stakeholder community,” the 2019 document stated.
The new 44-question survey, based on the JSP, intends to deliver on that statement. The survey serves as a self-assessment tool for medical device manufacturers, helping them identify their own medical device security maturity in areas like risk management, design control, structure, and governance. Use of the JSP is not required for survey participation, and companies using other maturity models can also gain valuable insights from the survey results.
Along with measuring the successes and shortcomings of the JSP, the survey will provide much-needed benchmark data on medical device security maturity. MDIC and HSCC are seeking one survey response per company or organization, and all responses are confidential.
“When manufacturers contribute to the survey, they will get a score that will help them to assess their posture in the sector,” Jithesh Veetil, program director at MDIC, explained in an interview with HealthITSecurity.
“And the learning, in turn, will help the industry, and also help us help the Public Health Sector Coordinating Council to update the JSP framework.”
Senior-level product security officers, risk managers, and quality managers were encouraged to complete the survey based on their working knowledge of their organization’s security posture and product portfolio.
“Cybersecurity risk is also a potential patient safety risk. It’s about protecting patient safety. It’s about protecting patient privacy,” Rob Suárez, CISO at BD and chair of the MDIC cybersecurity working group, told HealthITSecurity.
“That is really the reason why we want to give medical device cybersecurity this level of attention.”
RELATED
In this blog, we recap the “Understanding Integrated Risk Management for Medical Device” webinar.
Companies involved in developing medical devices understand the importance of risk management, but their approaches can vary significantly in terms of the time it takes to manage risk, the ability to connect risks to specific requirements and tests, and the capacity to pull together relevant documentation for an audit. To meet these challenges, medical device developers need a comprehensive approach to risk management.
In this presentation, industry and solution experts will explore how teams can integrate risk-based thinking into their product development lifecycle.
Attendees will learn more about:
Risk management in the medical device industry
Guidance and best practices to follow
How to manage risk analysis
The importance of risk traceability throughout project activities
Below is an abbreviated transcript and a recording of our webinar.
Understanding Integrated Risk Management for Medical Device
Mercedes Massana: So today we’re going to talk about risk management. First, we’ll start with the basics, the things we need to know to understand risk management, then we’ll talk about the elements of a risk management process, about some risk management tools that we can use, and then we’ll end with risk management and incorporating that into your traceability matrix.
So let’s start with the basics. So what is risk management? It’s the systematic application of management policies, procedures and practices to the task of analyzing, evaluating, controlling and monitoring risk. And in this case, we’re talking about product risk, not so much project risk, right? So all medical devices carry some level of risk, no matter how simple they are. There’s always some level of risk for the medical device, and we need to consider who can be hurt by the medical device. Who does this risk apply to? And that can be obviously the patient, but it can also be the operators or clinicians, right? The nurses. It could be bystanders, it could be service personnel working on the device. It could be even other equipment if we interfere with other medical equipment, and it could even be the environment.
Mercedes Massana: It is the responsibility of the manufacturer to determine how much risk they’re willing to accept, or the market is willing to accept for the intended use of the device. So the regulatory agencies don’t tell you what is acceptable from a risk perspective, but it’s up to the manufacturer to determine that.
So why do we practice risk management? Well, first of all, it’s so that we can produce safe products and release only safe products, right? So we want to prevent safety-related problems in the field. Having to recall product is very bad for companies, right? There have been companies that have gone out of business because of safety issues in the field. Having a good, well-documented risk management file can substantiate due diligence if somebody tries to sue you, so you have the documents that can help support that you did the right things.
It can also encourage a defect-prevention mindset. So when you start practicing risk management early on in development, you start designing with defect prevention in mind. You want to prevent defects that can cause harm and risk. It helps you identify potential safety issues early while you can still influence the design, right? And then, from a regulatory perspective, documents from your risk management files are always needed for submissions, and in audits, most likely these documents would be presented in audits.
And then it also allows risk-based decisions to be made throughout the product life cycle. So we think of risk management just as the product and things we need in order to get regulatory approval or to have in an audit, but really, having a robust risk management file can help us make decisions and verification, validation in manufacturing, even for our suppliers and what controls we ask them to implement. So having a robust risk management file can really help us in every facet of product development.
Mercedes Massana: So compliance is a big part of risk management. ISO 14971 is the application of risk management to medical devices. It is an FDA-recognized standard. It’s actually even called out in a couple of guidance documents from FDA, and it is referenced by a number of IEC standards. So we need to be compliant with ISO 14971 in order to get through FDA, and in order to achieve the CE mark. ISO 13485 mentions risk management 15 times, and it says that we must consider risk in supplier controls, for verification, for validation, in testing and traceability, for CAPA, even for training of personnel.
So this tells you how important risk management is to having a medical device, developing a medical device, and maintaining a safe device in the field. So risk management should be practiced first as a system-level activity, so we should start risk management from the top down. That means that very early in development, when we start our design efforts, we analyze the risk that the system can perform, just by knowing the intended use. We don’t even need to have a design. Then we attempt to mitigate those hazards and we drive risk controls through requirements that then get implemented in our design, so only the system can actually cause a hazard. The system might have many components, but unless I have all of the system put together, I can’t cause a hazard.
Design Transfer: Best Practices for Translating Your Device Design into Manufacturing Specifications
Manufacturing specifications are successful when they result in your medical device being produced consistently, over and over, and meet requirements and expectations, including those of design intent and quality specifications. In this blog post, I’ll share the best practices I follow when translating a medical device design into manufacturing specifications, specifically the drawings and specifications of parts and assemblies.
1: Start early
When thinking about the typical medical device product development paradigm, design transfer is usually depicted as one of the steps right before commercial production. Sometimes it is shown in parallel to design validation, and always after detailed design and engineering and design verification.
However, in reality, design transfer, especially the translation of your device design into manufacturing specifications, should start earlier.
Creating the right manufacturing specifications starts in the detailed design and engineering phase of product development. Manufacturing specifications are the drawings, part specifications, work instructions for assembly, testing requirements, and other elements of the Device Master Record (DMR), or the ‘recipe’ as I like to call it, to be able to produce your product. Thus, it’s key to be already deciding which technologies will be used to produce your parts and using a Design for Manufacturability (DFM) process to make assembly easier and more efficient and reflect those decisions in your drawings and specifications. And like the product development process itself, translating your device design is iterative in nature. Starting in the detailed design and engineering phase allows you to follow the steps further defined below.
2: Involve your manufacturing partners
This is the time to involve your manufacturing vendors for their input as to how to specify the custom parts and assemblies they will be supplying. They are the most knowledgeable regarding the tolerances and limitations their processes can generally provide while you, as the final device manufacturer, are the expert on the final design intent and criticality of the parts or assemblies they are providing. Working together will result in drawings and specifications that can be manufacturable and meet design intent. This is also another good time to incorporate other Design for Manufacturability (DFM) aspects for the largest positive impact to the design.
Translating your design specifications into manufacturing specifications is not a time to omit incorporating risk. Reference your risk analysis to determine which parts have functional and safety implications and tailor the specifications, including the quality specifications accordingly.
4: Remember the quality specifications
Speaking of quality specifications, having a strong quality control plan, based on risk, is also part of a successful translation of design specifications into manufacturing specifications.
Elements of a quality control plan include quality agreements with vendors; part validation expectations; various inspection requirements, including during receipt of first articles, incoming, in-process, and final acceptance testing; and ongoing process monitoring. Aligning expectations with suppliers of critical components is key. I see these quality specifications as partners to the part and assembly drawings and specifications.
Inspection testing is a good example to see the relationship from a design input specification to a manufacturing specification, specifically in this case, to an in-process testing specification. Say your device has a design input specification for a minimum flow efficiency of 80%. The 80% is based on the clinical need of the device. During design verification, the design is tested with a statistically relevant sample size to have an efficiency of 95% ± 2%. This range is also measured during process validation. Thus, in-process testing can be set at an 89% minimum. Note, this is intentionally tighter than the original design input, so that process issues and drift can be detected earlier.
Another example is related to sterilization. Say the design input is for a sterile product to have a one-year shelf life, i.e., the sterile barrier must maintain its integrity for one year. In this case, a Tyvek heat-sealed pouch is selected as the sterile barrier. During detailed design and development, the specific pouch is selected, the parameters for sealing are determined, and the corresponding peel strength of the seal is measured. Then design verification testing verifies that the pouch, when sealed under the selected parameters, does indeed maintain its sterile barrier after one year (accelerated aging). At this point, the corresponding peel strength of the seal can be used as an in-process specification to monitor the sealing performance. This minimum force specification is based on the performance data measured to date (typically from process validation), with adjustments to accommodate the observed manufacturing variability. Thus, the design input to have a one-year shelf life of the sterile product is translated into a manufacturing, in-process testing specification of a minimum peel test force of the seal.
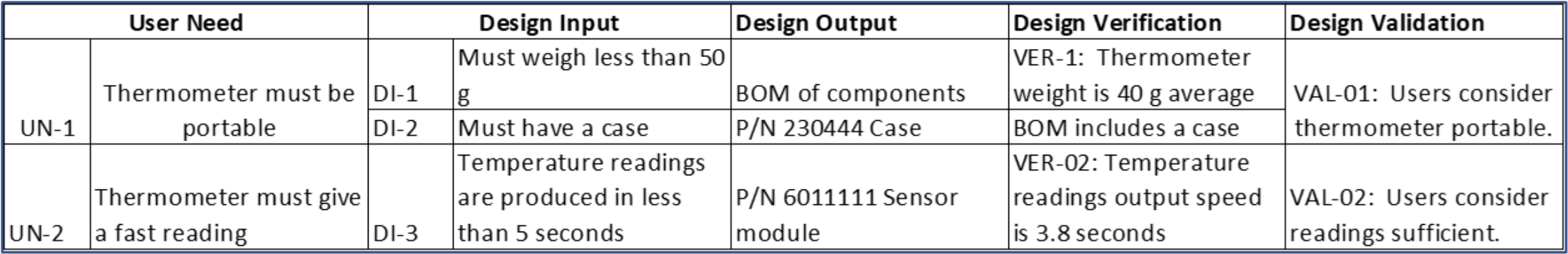
The FDA and other regulatory agencies expect a trace matrix associated with your medical device to show linkages from User Needs through Design Validation.
In the trace matrix, it’s where you can see the direct linkages between part and assembly (design outputs) specifications to corresponding design input specifications. Note that it need not be a 1:1 relationship. Multiple part/assembly specifications can be linked to a design input, and one part/assembly specification could fulfill multiple design inputs.
Here’s a snippet of a trace matrix for a fictional home-use thermometer. Especially for more complex medical technologies, a requirements tool such as Jama Connect® makes it more efficient to create and manage a product’s traceability matrix and ensure there are no gaps. As the Design Output column are manufacturing specifications, this is where you can see the traceability between the design input specifications to manufacturing specifications. It’s in this column where the manufacturing testing requirements in the examples above would be listed.
And a well-specified trace matrix is a good tool to use to understand the impact of future design changes, both for changes that occur after the design has transferred to manufacturing, as well as for any changes that may result from design verification and design validation. At times, if the risk is appropriate, you can choose to perform some design verification activities and design validation activities in parallel. Test units for design validation must be production equivalent, thus being able to trace which revisions of the design and resulting manufacturing specifications used units for design verification activities and design validation activities is important and to be able to justify the impact of any differences.
There are many activities to consider as part of manufacturing transfer. These best practices focus on translating the device design into part and assembly drawings and specifications, including the quality specifications, that are a part of ensuring your device is made the right way every time.
RELATED
Jama Software is always on the lookout for news and content to benefit and inform our industry partners. As such, we’ve curated a series of articles that we found insightful. In this blog post, we share content sourced from Medical Device and Diagnostic Industry – Celebrating the 2022 Medical Design Excellence Awards Winners & Finalists – which was originally published on April 8, 2022, by MDDI Staff.
Celebrating the 2022 Medical Design Excellence Awards Winners & Finalists
The medtech industry’s premier awards program honors significant achievements in medical product design & engineering that improve the quality of healthcare delivery & accessibility.
Since its inception in 1998, the MDEA program has honored significant achievements in medical product design and engineering that improve the quality of healthcare delivery and accessibility. More than just a beauty contest, the MDEAs convene an independent panel of esteemed jurors—made up of clinicians, engineers, and designers—to select up to six finalists, as well as Bronze, Silver, and Gold winners in each of the 10 product categories, along with overall Best-in-Show winner and Readers’ Choice. The annual competition recognizes products that are moving the $450 billion medical device industry forward through life-saving innovations and remarkable technological advancements.
Entries are scored based on five criteria: design and engineering innovations, user-related innovations, benefits to overall healthcare, benefits to patients, and differentiation in the market.
“This year’s entrants were amazing; each playing a significant role in changing the direction of the industry and bringing viable new innovations that overcome today’s most pressing challenges in the healthcare field,” said Daphne Allen, editor of MD+DI. “Congratulations to our winners and finalists who are shaping much-needed progress in a meaningful and impactful way.”
2022 MDEA Winners are as follows:
Best in Show: HemoScreen (PixCell Medical) MD+DI Readers’ Choice: CT in a Box (GE Healthcare)
Cardiovascular Devices
Gold: TYRX Absorbable Antibacterial Envelope (Medtronic) Silver (tie): HydroPICC and HydroMID (Access Vascular, Inc.), TELLTALE Electrosurgical Guidewire System (Transmural Systems) Bronze: ASSURE Wearable Cardioverter Defibrillator System (Kestra Medical Technologies, Inc.)
Digital Health Products and Mobile Medical Apps
Gold: Minuteful Kidney Test (Healthy.io) Silver: Oregon Capacity System (GE Healthcare) Bronze (tie): Bigfoot Unity Diabetes Management Program (Bigfoot Biomedical), FreeStyle Libre 3 (Abbott)
ER and OR Tools, Equipment, and Supplies
Gold: MOLLI (MOLLI Surgical Inc.) Silver (tie): Baxter EASYGRIP FLO41 (Baxter), Hugo Robotic-assisted Surgery System (Medtronic) Bronze: TN-Advanced Tibial Nailing System (DePuy Synthes, the Orthopaedics Company of Johnson & Johnson)
A complete list of finalists in the 2022 MDEA competition can be seen on MD+DI’s website.
The MDEA program accepts entries worldwide from companies and individuals involved in the design, engineering, manufacture, or distribution of finished medical devices. The competition is open to finished medical devices, including instruments, machines, implants, in vitro reagents, mobile medical applications, or other related products that are intended for the diagnosis, cure, mitigation, treatment, or prevention of disease or other conditions in humans or animals.
In this blog, we present a research report conducted by Axendia, a leading Life-Sciences Analyst firm, and presented by Jama Software, we walk through groundbreaking new research about the costly impact of ineffective requirements management in the medical device industry.
Axendia Report: The Costly Impact of Ineffective Requirements Management
Medical device organizations are continuing to sharpen their focus on developing high-quality medical devices aimed at improving patient outcomes. However, many struggle with effectively managing requirements and traceability across the product development lifecycle. This can be costly, risky, and lead to delays in new product introductions when considering the increased complexity in medical products, competition, and the regulatory landscape.
In this research report conducted by Axendia, a leading Life-Sciences Analyst firm, and presented by Jama Software, we walk through groundbreaking new research about the costly impact of ineffective requirements management in the medical device industry, including:
The impact of having an ineffective closed-loop requirements management process.
The critical importance of requirements management to achieve improved patient outcomes, product quality, and time to market.
The negative impact on budgets, traceability, verification, and validation activities when relying on manual processes
Axendia is a leading analyst and strategic advisory firm focused exclusively on the Life-Sciences markets. They provide strategic advice Business, Regulatory and Technology issues and trends enabling our clients to prepare for, adapt to, and overcome disruption.
In this post, we recap a recent webinar hosted by Jama Software on the topic of requirements management in the medical device industry
Requirements management solutions enable the unification of siloed processes and data that often reside in outdated, disparate, and disconnected legacy systems. However ineffective and inefficient, the industry still relies overwhelmingly on static documents housed in Excel/Word and relies on manual processes that add significant risk to the development process.
Axendia conducted a research study focusing on the medical device industry’s approach to requirements management with a goal to identify and analyze the drivers, barriers, trends, and value of requirements management across the product development lifecycle.
9 out of 10 (87%) Executives surveyed for this report admitted to having a ‘not effective or somewhat effective’ requirements management process.
In a recent webinar, Axendia’s Senior Industry Analyst, Sandra K. Rodriguez and Jama Software’s medical and life science principal solutions lead, Steven Meadows, shared the outcomes of the research including:
The impact of having an ineffective requirements management process
The critical importance of requirements management to achieve improved patient outcomes, product quality, and time to market
The negative impact on budgets, verification and validation activities when relying on manual processes
Requirements Management in the Medical Device Industry
Sandra Rodriguez: Thank you for that introduction, Marie, and good morning, good afternoon, or good evening, depending on where you’re joining us from today. Really quickly about Axendia, we were founded in 2005. Our headquarters are in Philadelphia, Pennsylvania. I’m physically located in San Juan Puerto, Rico, it’s a balmy 84 degrees today. What’s unique about Axendia is that our analysts all have industry experience. And combined, we average about 25 years of experience. We work with startup companies as well as fortune 500 global clients. And we really focus on the intersection of the business regulatory and technology trends that impact the industry. So really looking forward to sharing the outcomes of our latest market research on the state of requirements management in the medical device industry.
Just really quickly too, for the folks of you on the webinar today, congratulations, you will be the first to receive a copy of the research report. I’m not going to cover the report in its entirety today because we don’t want to take away the thunder of it, but we do hope that you will find today’s information valuable and timely and that you get some great takeaways from the report. Before I go into that, though, just a quick acknowledgment that the content of today has been sourced from our quantitative and qualitative research, as well as our interaction with FDA’s officials and industry executives, and then some firsthand experiences from our clients as well.
All right, so let’s start with the demographics. We surveyed a multitude of companies from around the world, companies that perhaps make more than one type of medical device product, but here you can see that the majority do market and sell single-use disposable consumable devices followed by diagnostic devices that have both hardware and software components and software as a medical device.
So those were the top three. As a result of that, we wanted to look at the data in a little bit more kind of slice it and dice it. So we specifically picked out software as a medical device, and you’ll see in the report, and as well as in the presentation today, how the opinions vary based on the type of medical device company that we surveyed for this report.
In addition, we got a good mix of small companies and large companies. The majority of the companies that did respond to the survey were under 50 million. So they could be startups, they could be a little bit pre-market followed by the $1 billion to $5 billion size companies when it comes to their revenue. So another thing that we did was we went ahead and compared these survey responses based on those two different size companies. We also got a significant number of R&D and product development personnel to take the survey, which is important.
These are what I call the boots on the ground, the companies that really understand requirements management inside and out. They’re the ones who are working on the new products as well as quality assurance personnel. And then we had a 17% representation of executive management. So another thing that you’ll see in the report is that we went back to the data and did do some comparison and filtering based on these three personas. And we were really surprised to see how the attitude shifted.
Sandra Rodriguez: From a geographic standpoint, we had a really good representation. Overall, the majority of survey respondents do work for companies that sell in market products in North America, so Canada, the United States, and Mexico followed by Europe and all the EU member states. And then, of course, Asia, South America, Africa, and Australia. So again, a really great representation of the medical device industry.
So this was our first question. And following the demographics, I think it’s really important to point out that this was the first question that it came when it came to requirements management processes. We wanted to understand from the market standpoint if people felt that their organization’s requirement management process was either effective or I’m sorry, not effective, somewhat effective, very effective or ideal. We were really surprised that when you combine not effective and somewhat effective, straight out the gate, we have a 68% of the market saying that there is a lot of room for improvement here. This is what I call aiming for average. And it’s definitely time for this to change because the complexity of the devices is changing. The regulatory landscape is changing. We have a lot more software as a medical device coming out there into the marketplace. And there’s a lot of hardware and software components that are going into these products, whether you’re doing remote monitoring of the products once they’re out in the field or in the patient.
So because of the complexity alone, you really need to have… You would want to see the very effective and ideal numbers go up because when we combine them, we’re only at 31%. And what’s really shocking is that only 2% of those companies that we surveyed for this research project believe that they have an ideal process. So we wanted to take a look at those numbers from the different personas. The quality personnel were of a little bit more positive. They believe that their requirements management process is very effective or ideal. So they account for about 53% of that. But when you look at the executive management, to have 87%, so almost 9 out of 10 executives indicating that their organization’s requirement process is not effective or only somewhat effective, that’s pretty shocking. Keep in mind that these are the folks that own the budgets.
So if you know that there’s a problem, we really need to do something to incentivize these folks to make the necessary change and get to that very effective or ideal state, even on the R&D and product development side, the same holds true with 68% of those professionals saying that their organization’s process is somewhat effective or not effective.
So we also asked the closed-loop system question. And we define a closed-loop system as one in which the desired output depends on the input signal and the feedback elements that are going to enable end-to-end traceability. So when you’re looking at a typical product development cycle, you have the finished product here in the middle. So you’re going from concept to prototype. Clinical, if you need to have a clinical trial for the type of medical device that you’re looking to bring to the market, going into manufacturing, marketing, commercial, and then obsolescence.
Sandra Rodriguez: So having that continuous feedback loop, really closing loop around that system, and having the necessary traceability that’s going to be required from a quality and from a regulatory standpoint. So we ask them, how effective is your organization at closing the loop across the product development cycle. Now, mind you, this was question number two. And we see here that 68% of the market, again, admitted that that process is not effective or only somewhat effective.
So again, really need to make the necessary changes there, and probably invest in the solutions so that you can close the gap and get that traceability and close the loop across the product life cycle. It’s interesting as an analyst because we don’t sell or implement software, but we stay really close to the market. And we follow these trends and we see significant investment in the life sciences when it comes to digital transformation or investing in business systems.
But it’s important to point out that without a product you wouldn’t be in business. So the solutions and the systems, the tools that you have in place when it comes to your product should be as important as the systems and tools that you give your salespeople, or your ERP, or however, you’re investing in those systems. You really need to make the necessary investments in order to make sure that your product is of the highest quality and that you can get to market sooner and on time and on budget because we’re going to learn in this presentation that the industry is really struggling with that.

![[Webinar Recap] Understanding Integrated Risk Management For Medical Device](https://www.jamasoftware.com/media/2022/07/2022-07-12_Understanding-Risk-Medical-Webinar-Social-Image.png "[Webinar Recap] Understanding Integrated Risk Management For Medical Device")




![[Webinar Recap] The Costly Impact of Ineffective Requirements Management in the Medical Device Industry](https://www.jamasoftware.com/media/2022/02/2022-02-07_Azendia_Blog-Social-Image.png "[Webinar Recap] The Costly Impact of Ineffective Requirements Management in the Medical Device Industry")