In this post, we pull out key takeaways from a recent whitepaper written in conjunction with Beanstock Ventures on the new EU medical device regulations (EU MDR) and how they might impact medical device development.
As medical device technologies have rapidly advanced in recent years, regulations governing definitions, testing, and post-market activities have struggled to keep up. The pace of change and adoption of these technologies has made it difficult for governments and agencies to create the kind of inclusive and expansive rules that will ensure safety.
In response to this expanding market, the European Union released new guidance governing medical devices. With the release of Medical Device Regulation (MDR) 2017/745/EU, in 2017, the EU has issued the first updated regulations in more than 20 years. The new Medical Device Regulation (MDR) 2017/745/EU addresses software as a medical device [SaMD], as well as other products. It also places stringent requirements for compliance with post-market activities and post-market surveillance. While enforcement of these new regulations was scheduled to begin in May 2020, it was postponed until May 2021 due to the COVID-19 pandemic. What do these new regulations mean for the medical device industry? Experts from Beanstock Ventures explain what you need to know for EU MDR compliance.
The EU Medical Devices Regulation (MDR) has replaced the EU Medical Device Directive effective 26 May 2021.
The EU MDR is greatly expanded to cover more devices, including Software as Medical Device, implantable devices, contact lenses, and many digital health technologies. It also promotes a lifecycle approach to regulation.
EU MDR requires improved device traceability by introduction of a unique identification system, or UDI (see section 05), for medical devices approved for use in the EU. To keep track of devices through every lifecycle stage, a device identifier (UDI) will be assigned, and all production series will be marked with a production identifier.
EU Medical Devices Regulation (MDR), adopted by the European Parliament and Council as REGULATION (EU) 2017/745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017, has replaced the former EU Medical Device Directive (MDD) and went into effect 26 May 2021. After this date, the MDR is applicable for all medical devices sold (developed or imported) in the European Union.
The most important changes in the EU MDR include:
Increased scope of medical device definition;
New classification rules (including Rule 11 that specifically addresses software);
Increased scope of general safety and performance requirements, technical documentation, and clinical data and evaluation requirements;
Introduction of traceability and identification system and database; and
The medical device market in Europe is growing fast and is expected to reach $61.4 billion by 2025, up from $48.9 billion in 2020. A large aging population, demand for more surgical procedures, and technological expansion are driving this trend.
The Medical Device Directive (MDD) is a legal framework that includes three directives that are designed to regulate the safety of medical devices in Europe. The MDD came into effect during the 1990s; however, since that time, the technology landscape has quickly evolved. Many technologies that didn’t exist previously, such as software as a medical device (SaMD), are now widely used to treat, diagnose, mitigate, and even prevent disease.
Governing agencies were concerned that the MDD was outdated and didn’t address many evolving safety threats. As a result, they developed a whole new set of standards called the European Medical Device Regulation (MDR). Medical device companies that are already compliant with MDD regulations shouldn’t be fooled into a false sense of security. The EU MDR is far more comprehensive than the previous regulations were. Understanding what the MDR is, changes to the regulations, and what to expect can help your company get prepared and stay compliant in the future.
The Medical Device Industry is Undergoing a Time of Rapid Growth
The industry in Europe is expected to reach $61.4 billion by 2025, up from $48.9 billion in 2020. The rules that applied to medical devices are shifting to keep up with that growth and the resulting innovation.
What is the EU MDR?
The European Medical Device Regulation is an entirely new set of standards that outlines rules around the production and distribution of medical devices in Europe. The regulations were designed to ensure that companies produce medical devices safely and that potential risks are mitigated. The prior document, the MDD, had roughly 60 pages, compared with the MDR which spans 174 pages. The new document includes a 13-page introduction, 123 articles, and 17 annexes. Additionally, there are 42 implementing acts, which further clarify the MDR, and 12 delegated acts, which modify and amend the regulation.
Understanding the major changes can help you better understand how to stay compliant.
MDD vs. MDR: What’s the Difference?
MDD REGULATIONS: Roughly 60 pages in length and focused heavily on the preapproval stage of medical device management.
MDR REGULATIONS: Includes 174 pages that present a lifecycle approach to regulations. Expanded to include new categories of coverage, such as cosmetic devices, contact lenses, and many others, as well as more rigorous oversight.
To learn more about EU MDR and get a better understanding of the changes being made, download our full EU MDR eBook.
Editor’s Note: This posts on EU MDR was originally published here by MedTech Drive and written by Greg Slabodkin.
Dive Brief:
While the Medical Device Regulation’s May 26 go-live date marks a significant milestone, MedTech Europe warned in a statement that MDR challenges remain, limiting the industry’s ability to “seamlessly supply certified devices under the new rules.”
The European trade group contends that despite the EU MDR going into effect, “some key pillars” of the necessary infrastructure are “still not fully operational or even in place,” creating challenges in particular for many small and medium enterprises.
CEO Serge Bernasconi argued that the complexity of MDR and the delay in the new regulatory system’s full readiness is resulting in European patients “losing their previous opportunities to be the first to benefit from critical medical technology innovation.”
Dive Insight:
MedTech Europe had been sounding the alarm for some time about MDR’s potential to disrupt product supply due to issues around the ability of the EU’s infrastructure to ensure a smooth transition when the rules came into force on May 26. Now that the new regulatory regime has reached Wednesday’s date of application, the trade group is once again warning that significant challenges remain unresolved that could negatively impact the sector.
A range of MDR uncertainties and potential problems are hovering on the horizon. While the medical device industry has resolved some near-term pressures as the delayed landmark regulation goes into effect, MedTech Europe on Wednesday listed a litany of MDR challenges that are hindering the successful deployment of the new regulatory system.
These MDR challenges include:
Non-harmonized interpretation and application of MDR rules across the EU.
Limited capacity among notified bodies, especially for certification of new and innovative devices.
Uncertainties with regards to pending discussions on the rules and agreements between the EU and other countries such as Switzerland.
Unpredictable recognition of MDR certifications at the international level vis-à-vis regulatory approvals from other jurisdictions.
“Such challenges need ongoing attention and work by the EU Commission and Member States, if Europe is to ensure a workable system in the long-term,” MedTech Europe said.
The trade group also highlighted an urgent need for attention to be given to the In Vitro Diagnostic Regulation, a major overhaul of the diagnostics sector slated to go into effect in 12 months.
“As with the MDR, the medical technology industry fully supports the new regulatory regime for IVDs but due to many factors, the system is not yet ready to support its implementation. Urgent solutions are needed here as well, and lessons learned from the MDR implementation should be taken into account,” according to MedTech Europe.
The group added that it is working with the EU and stakeholders to “rapidly propose solutions to avoid disruptions” in supply of medical devices and diagnostics.
2020 has been a year that’s been described as “unprecedented” and “unparalleled” – as well as other descriptors probably best left out of our blog. As we close out this year, it’s hard to say what awaits us in the new one. One thing that we can be sure of is that innovation in medicine, science, and technology shows no sign of slowing down.
As we enter a new year of technological advancements, Jama Software asked select thought leaders – both internal and external – across various industries for the trends and events they foresee unfolding over the next year and beyond.
In Part I of our four-part series, we ask Steven Meadows, Medical Device Solutions Lead at Jama Software, and Founder & CEO, Shawnnah Monterrey at Beanstock Ventures to weigh in on trends they see in the medical device industry for 2021.
Note: Now that our 2021 Predictions Series is complete, you can also go back and read, Part II,Part III, and Part IV.
What are the biggest trends you’re seeing in the medical device industry right now? How will they impact products, systems, and software development?
Steven Meadows:
From a product perspective, there are a number of areas in the medical space that are set to grow in 2021 and beyond, including robotics nanotechnology, wearable health tech, and extended reality devices.
With more medical devices incorporating software components and connecting to systems, like hospital networks, enhanced cybersecurity is becoming increasingly important. Medical device companies will continue to invest heavily in this area through 2021 and beyond.
Shawnnah Monterrey:
There are three primary trends that I am seeing: A shift in initial product launches from Europe to the United States; An increase in Software as Medical Device products; and an increase in IVDsfrom lab-developed tests.
We are seeing a shift in initial product launches from Europe to the United States, due to the upcomingEU MDR regulations.Typically, US–based companies would launch medical products in Europe first, followed by a US launch shortly after due to the rigor and lag time in receiving FDA approval. CE Mark was typically more straightforward, required less clinical evidence, and was a faster process, especially when companies could self-certify.EU MDR is currently being perceived to be more onerous than the FDA regulatory clearance process and is therefore creating a trend of product launches occurring first in the United States and then in Europe. The cascading impact would therefore be the need for more services providing expertise in regulationsto support EU MDR and a need for a more harmonized regulatory strategy.
The second trend we are seeing is an increase in Software as a Medical Device (SaMD). There have been numerous healthcare, fitness, and consumer products launched into industry recently that were not previously considered a medical device. As a result of the SaMD FDA guidancecombined with the EU changes from CE, MDDto EU MDR, many products that were not originally deemed clinical products or medical devices are now falling into that category. Products that were previously not classified as medical deviceswere not necessarily held to the same standards thatare required to produce a medical-grade product such as quality management, design control, risk management, clinical studies, and post-market surveillance. This shift will cause companies to delay product launches in order to satisfy the regulatory requirements, resulting in an increased need for training, systems, software, and tools to make it easier for companies to make that transition. Since these companies generally lackquality and regulatory expertise, additional support will be requiredto deliver medical-grade software products.
The last trend we are seeing is an increase in IVDsfrom lab-developed tests (LDTs), research use only (RUO) and emergency use authorization (EUA).There are numerous laboratory-basedtests and associated software that havebeen developed to support very complex assaysthat are being used clinically which has recently been deemed outside of the scope of LDT guidance.Products that were covered under CLIA are now required to be cleared with the FDA and EUas an IVD.From a CLIA perspective, the focus is on validation. Validation is centered on the intended use of the product and workflow. But with medical devices such as IVDs, there needs to be a shift to bringing it down to verification levels –namely design verification. Design verification is quite different than validation, design verification islooking at the design aspects of the product, not from a workflow perspective, but from a design feature perspective.Design verification occurs at the subsystem and system level, where the subsystem is the breakdown of the system from the architecture.The scrutiny will be from a hazard perspective for the entire system and a failure mode analysis perspective at the subsystem level. Finally, ensuring that there is a full product definition and full verification associated with theproduct. The impact is that it is going to require taking a different look at the software that has been provided and looking at it differently from a process software perspective to an IVD perspective this might require looking at the architecture and redesigning the system to break apart the items that were allocated to CLIA and the lab system processes versus the actual product that performs the clinical interpretation and the diagnosis.
An overarching impact will be evolving medical device productdevelopment from a methodology perspective. Manycompanies are lean and agile and are performing continuous integration and development of their products. It will be important to set up processes and procedures to still allow for that while continuing to meet the regulations. From a systems perspective, it would be beneficial to take advantage of available tools.There isa myriad of tools in place that can assist indevelopingquality management systems, manage design control, support requirement and risk management, testing, test automation, and deployment. There is anopportunity for companies to start investing in tools to help withcompliance and save time and money whilestill maintainingtheir efficiencies.
In terms of product and systems development, what do you think will remain the same over the next decade? What will change?
Steven Meadows:
Software is becoming an increasingly used component in medical devices and we expect this trend to continue. More of our customers are adopting an Agile methodology when developing their software, a shift that has been happening for some time, and will continue to over the next decade.
Shawnnah Monterrey:
Over the next decade, I see the use of Artificial Intelligence (AI) supportingthe creationof sophisticated data-drivenalgorithms such as autonomous diagnosis. AI is gainingrenewed traction, even though it has been used in medical products since the 2000s. I believe there will continue to be a tremendous amount of investmentin the development and understanding AI in the healthcare space and how it could be used to improve clinical outcomes. AI will continue to progress over time as the industry develops an understanding of the utility and how it can contribute to guiding clinical decisions as an autonomous tool. Thereis FDAdraft guidance around AI, however, I think the biggest potential constraint will be how to continuously improve upon the algorithms as new data comes in, without requiring a new regulatory submission.
The other trend I see progressing over the next 10 years is one we are already seeing,cybersecurity. This trend is a bi-product of the increasing numbers of clinical cloud applicationsthat integrate with or use medical devices. As the need for sharing clinical data, advanced computing, remote diagnostics,and accessibility increases, so will the migration of healthcare applications to the cloud.This increased need will be the catalyst that pushes up the demand for securing patient data not only in terms of patient privacy but also in terms of patient safety.We have seen several instances now where, established medical device manufacturers, medical device cloud software, could be hackedby an unauthorized person and potentially cause adverse side effects to the patient. Such hazards could trigger a class I recall, the most serious type of recall. Not only do these hazards put patients at risk, but they also impact a company’s reputation and financial viability. Occurrences like this will justifiably result in a growing emphasis on creating software systems, processes, and tools tosupport the industry inmaturing standards and guidance around cybersecurity.
In terms of what will remain the same in the medical device industry throughout 2021, I would say that the regulationswill remain unchanged. It is unlikely we will see a change in this coming year. To the contrary, we will likely see an increase in the regulation. Ipreviously discussed the potential for harmonization. However, I do notbelieve that will happen until there is a push for it or until the FDA acts on the overlap between many of the guidance documents. Additionally, there will need to be harmonization on the development methodology across different devices so there is not a need to have different guidance documents. In short, I do not see the regulations changing over the coming year.
In discussing 2021, we should also take a moment to discuss Covid-19. It’s not my belief that things will change from a Covid perspective. The trend that I see continuing involves businessesincreasingly going digital, putting pressure on products to focus on cybersecurity. Specialties, such as telemedicine will likely increase, requiring true patient authentication and there will be progress, but a delay in medical device development in areas of selected surgeries.
Regarding development teams, I think Agile is here to stay. I thinkthat development teams will continue to use the agile approach and methodology. The organizational struggle of internal alignment of the quality management system will persist until there is adisruption in the regulatory environment itself. Relative to the need for systems as software tools become more useful and more integrated. The main area I see increasing are the new entrants into the market.There has been a large amount of new investment and I believe that will continue.I think we can assume that there will still be quite a bit of investment opportunities for new players.
What sorts of process adjustments do you think development teams will need to make to accommodate these changes? Do you think they’ll need to make technology investments, process adjustments, or both?
Steven Meadows:
As we continue to deal with the new normal, I expect medical device teams to continue to invest in collaborative product development software, enabling their teams to continue bringing new devices to market in a safe and competitive way. Development teams, using legacy product development platforms and Word/Excel need to switch to a modern alternative, in order to be as successful as possible in 2021.
Shawnnah Monterrey:
My belief is that development teams are going to need both technology investments and process adjustments to accommodate the changes that are coming. There will be a heavy reliance on technology as the software realm moves more towards focusing on core IP and leveraging the existing software components available. For example, there could be an organizationsuccessfully providing cybersecurity packages where other organizations may not have that expertise, so they would be leveraging assets from another organization.In short, software re-use will be important.
My sense is that the industry will shift more towards investing in technology such as AI and mechanisms to improve data sharing.There is an opportunity to really understand the clinical value that the data brings.Currently, the healthcare environment iscomprised of disparate systems that will eventually need to come together to be able to fully leverage the data and use the data in unique yet meaningful ways.
We are currently seeing a big shift where the medical device companies and their quality management systems do not satisfy the way in which engineers perform medical device development. For example, software engineers are very agile and tool oriented by nature, while many established companies still utilizea paper-based quality management system or document control system. We’d like to see software teams and the Agile methodologiesdrive changes to the quality system so that the quality system can be nimbler and more adaptiveallowing them torelease compliant products faster.
We have observed a hindrance in the companies and auditors themselves to adopt a more progressive and nimble approach that is not necessarily driven by the regulations. Our interpretation is that the intent of the current regulations is to be nimble andstill provide safe and effective products.My belief is that if we do not make a change to the current way in which we develop medical devices in thecorporate environment, innovation from these larger organizations will be stifled. The innovation gap will ultimately be filled by startups entering the marketplace and experiencingimmediate success because the old way of doing things is no longer sustainable or effective. I foresee a trend in an increase in the volume of new, emerging, small players. These players are going to be nimble and will look at the regulations from a different perspective.
How do you foresee regulations shifting in the medical device industry over the next decade?
Steven Meadows:
Key medical device standards and regulations are constantly changing. Notably, EU MDR is undergoing a number of changes and will need to be fully implemented by May 2025. Key standards like ISO 14971 go through multiple iterations and Jama can help our customers stay current with these changes.
Shawnnah Monterrey:
I will say something that ‘should’ happen, not that ‘will’ happen,relative to regulatory changes in the medical device industry over the next decade.Ideally, there would be harmonization across the guidance documents. There are too many guidance documents and many of them are repeating the same requirements but in different ways. With a harmonization effort, it would remove the perceived burden of applying multipleguidance and standard documents. With the current format, there is a need to understand each and every guidance and standard document andto be able to apply them all correctly. With more alignment and more harmonization, it would facilitate adoption and push forward innovation.
The impact of harmonization would certainly be in facilitating development. Withone cohesive guidance document and less confusion in applying multiple regulations and guidance documents across development, it would result in more development. If there was more alignment across the agencies to create cohesive processes, it would be easier to get consistency in the application of it. Additionally, the FDA’s time to approve products would be reduced because the FDA would be seeing similar submissions and be able to focus on the critical aspects of the product.
Anotherregulatory change that I expect to grow over the next decade is with the FDA’s adoption of third-party review organizations. The FDA has begun utilizing third-party vendors to streamline and offload their burdenof reviewing Class I & II devices. By working with approved partners to facilitate the review, the FDA can focus on higher risk devices. This allows a company’s submissionto get reviewed more quickly. Over the next decade, the FDA will likely become more sophisticated in this partnership with third-party organizations. The impact would be a faster time to market, possibly launching products months more quickly.
What do you think will be some of the differentiators between a company surviving to see 2030, and those that do not?
Steven Meadows:
Medical device companies that embrace a proactive approach to quality will ultimately find fewer issues with their products, improve customer satisfaction, and stay competitive for the foreseeable future. Companies that invest in best-of-breed medical device development solutions like Jama Connect will have the upper hand in reducing risk and complying with various standards and regulations.
New cutting-edge technologies in the medical field, like robotics nanotechnology, and wearable health tech devices, bring complexities for medical device companies and risk for patients and consumers. Jama Software will continue to serve as the leading solution and partner to help innovative companies bring medical products to market, in a collaborative, safe, and efficient way.
Will those trends still be prevalent 5 years from now? 10 years?
Shawnnah Monterrey:
I believe there will be disruption in the regulatory space in the next five to ten years, so I do not see the trends staying the same.
It will be important to have industry guidance by having industry experts partner with the FDA to help make those improvements. Based on the FDA approach currently, I believe they will welcome industry guidance.In relation to the previous discussion of the FDA 510(k) Third-Party Review Program, if we want to put it into a broader bucket, this is really the FDAutilizingexternal resources to support the industry.However, the collaboration is not simply due to a lack of resources, it is also because there is a very challenging, difficult infrastructure to navigate. I believe that there is a push to simplify the system and there will be a reliance on industry partners to do so. The industry partners will include those that are doing software product development, providing quality regulatory services, and providing tools to help facilitate medical device development.
What innovations in regulation in the medical device industry do you hope to see in 2021? 5 years from now? 10 years?
Shawnnah Monterrey:
The innovations in regulation in the medical device industry that I hope to see is the increasingopportunities for companies to collaborate with the FDA. I also hope to see more companies, larger organizations, or startups, take a more lean, nimble approach and be more receptive to the Agile methodology that software development teams are currently using. Current processes and procedures were generally derived from good engineering practices;however,perspectives of quality and regulatory experts and independent auditors remain the same. I would like to see them be more adaptive towards compliance,as compared tothetypical approach of checking the box, and instead, establish a partnership with their medical device development team to help facilitate compliance without sacrificing the quality of the product.
More specifically, I would like to see more efficient uses of tools and technology to support medical device development. There are many tools out there, butthey are very disparate.I believe that with more cohesion between those tools it would better facilitate medical device development to comply with the regulations.
In five years, my hope would be that there would be a first pass at simplifying the guidance documents.
In 10 years, I hope to see a streamlining of the processes. I hope for standardization of the submissions.I would hope to see a simplification of the current guidance documents across the board in the form of a standard method for supporting 510(k) and PMA submissions.
Stay tuned in the coming weeks for additional 2021 predictions! In the meantime, to see more information specific to the medical device development industry, we’ve compiled a handy list of valuable resources for you!
Editor’s Note: This post about getting the most out of your lab partnerships ahead up the upcoming EU MDR was originally published here on Medical Design and Outsourcing on October 5th, 2019, and was written by Joseph Tokos, Technical Director of Chemistry at WuXi Medical Device Testing.
Medical device manufacturers have a lot to keep track of these days. Between ongoing device development and preparing for the EU MDR, it’s more important than ever to maximize every partnership.
Outsourcing device testing to laboratories or contract research organizations (CROs) offers much needed relief to manufacturers and an opportunity to enhance EU MDR preparation efficiency. Here are a few insider tips to make the most of your relationship with your testing partner:
Don’t hold back on the details
Your testing partners need to ask a lot of questions about your device to be able to design a concrete test plan and accurate quote. It may seem like they’re asking for too much information before you even sign a contract, but these fine details can make or break a test plan. Failure to thoroughly detail device materials, for example, can lead to flaws in the extraction plan, such as the wrong solvent, equipment, temperature or extraction time. Complete and accurate dimensions are important because the extraction ratio will depend on wall thickness. Surface area information is also important because testing partners need to know whether the device will need to be cut into pieces for testing and whether there would be implications of doing so.
Providing all of the information upfront will save time and help prevent delays, denied submissions due to inadequate results and increased costs when studies aren’t designed properly.
Before requesting a quote, gather detailed information on the following:
Purpose, category and patient contact time
Size, thickness and surface area
Materials, colorants, pigments, adhesives, additives, polymers and manufacturing aids
Procedures used to manufacture and sterilize the device
Parts and composition
Existing data from previous testing
Read up on regulations
Understanding how new regulations and standards apply to your devices will empower you to educate your internal team and instill a sense of urgency to collaborate with your testing partner. Making an effort to keep abreast of the latest on EU MDR will allow you to have more productive and timely conversations about your test plans.
If you don’t have the bandwidth to dig into testing requirements — after all, that’s part of why manufacturers work with outside resources — you’ll at least want to check in with each person or department involved in device development. Design engineers and material suppliers, for example, will help you understand the intricacies of the device’s design, materials and parts.
It’s also important to note that patient contact time and in vivo testing methods will be under heightened scrutiny. Devices with shorter patient contact time are now going to be held to the same standards as longer patient contact time. This means the current data packages on even Class I devices may be incomplete moving forward. If the duration of patient contact is shorter than 30 days, be sure to ask your testing partner if you have remaining gaps in your submission that will require new testing. Also, take this opportunity to evaluate all of your options with existing in vivo device data or product family grouping data to minimize in vivo testing.
Learn from others’ mistakes
Sometimes, even the best intentions can’t guarantee a smooth testing experience. Beyond looking into what the regulations require and providing thorough product information, there are a few pitfalls that can lead to incorrect testing parameters, delays and added costs.
One common mistake is providing outdated information from a previous design. Start each device information request from scratch instead of copying and pasting from a previous form; then, investigate any gaps. This forces you to consider all the details carefully, which reduces the risk of an improperly designed test plan. Getting it right the first time reduces the risk of needing additional device samples (test article) and repeating tests.
It’s critical to provide truly representative test article of the devices produced by manufacturing. Using a prototype, for example, could yield inaccurate results and put your submission’s approval at risk because prototypes may be made of different materials or employ a different manufacturing process.
Another easily avoidable mistake is failing to describe any device changes in detail. Your testing partner can better address risks of your updated, next-gen device if they have a good understanding of what prompted the updates. Whether you addressed patient safety concerns or made updates to improve user experience, communicating the reasoning behind any device changes allows a lab to tailor the study.
Finally, don’t misunderstand the role of chemical characterization. While biocompatibility testing is important, chemical characterization is what identifies risks you didn’t even know exist.
The new regulation, which emphasizes patient safety, traceability, and transparency, will impact medical device developers, manufacturers, and suppliers of all sizes across Europe, and will require fundamental changes throughout the entire device lifecycle.
In order to better understand the regulation and its impact on our medical device customers, we sat down with Satyajit Ketkar, Principal System Architect & Engineer at Velentium, to learn more about what MDR is, what medical device developers can expect, and what they should be doing to prepare prior to the May 26, 2020 implementation date.
Jama Software: What is EU MDR and what can medical device developers in Europe expect?
SatyajitKetkar:
MDR stands for Medical Device Regulation. It combines and replaces the existing directives. Once the MDR goes into effect (on May 26, 2020), the existing directives will be obsoleted and invalid. All medical device manufacturers will be required to comply to the MDR. Right now, there are three different directives: The Medical Device Directive (MDD), the Active Implantable Device Directive (AIMDD), and the In Vitro Diagnostic Devices Directive (IVDD) — The EU governing body took two of those —the MDD and the AIMDD — combined them together and brought it to the next level to make it a regulation, which means that it’s extremely explicit and binding. There’s also the IVDDR, like the MDR and will replace the IVDD. The MDR is also harmonized with a lot of the latest standards. EU MDR is supposed to provide the medical device manufacturers a much more comprehensive regulatory guidance on how to manufacture a medical device. But it goes way beyond that, it goes from phase-zero feasibility, all the way through end–of–life (EOL) of the device. There’s a lot of emphasis on clinical, post-market surveillance, how to handle complaints, auditing, review cycle – whether it’s three-year or five-year. There’s a lot more content in there. It’s a pretty large document. It’s supposed to be in the same line as the data protection regulation (GDPR).
Jama Software: Why are they implementing new regulations around medical device development?
Satyajit Ketkar:
They’re basically pushing these regulations up to a level where the ambiguity and the ‘legalese’ is taken out. In the past, a directive was set out through the EU legislative body from Brussels, and then each competent authority for each country would interpret it in their own way. Each of the notified bodies, like BSI, TÜV, or DEKRA, had their own interpretation of the directive. So, if you’re a legal manufacturer, you could go to each one of these bodies and get a different implementation of the directive. What this new EU MDR regulation is supposed to do is to clear that up. Everybody now operates in the same manner, in the same language. And because of that, the regulation is large.Whereas the MDD about 60 pages and has 23 articles and 12 annexes, the MDR is about 175 pages and consists of 123 articles and 17 annexes.
Jama Software:
What are the top things that you suggest medical device companies do to prepare for the new regulation?
Satyajit Ketkar:
Overall, one of the first things I would recommend any medical device manufacturer do is to read the articles that talk about device classification and understand where they fit in.
If you’re an existing manufacturer of a product that’s already out in the market that falls under either the MDD or AIMDD, you’ll need to understand where you fall on the new spectrum. You must understand if your medical device still falls under the same classification, or if it has changed.
Most likely, they’ve gone up in classification. If they were Class 1R or Class 1M, now they’re Class 2A. If they were Class 2A, they’re now Class 2B. If they were Class 2B, they’re now Class 3. And of course, the current or future AIMDsremain as Class 3.
So, what does that mean? That means that the rigor of their development processes and the evidence that they mustmaintain, the DHF (Design History File) records and what they mustprovide has just gone up by quite a bit. When these directives were originally written back in the ‘90s, a lot of the technologies we’re working with today didn’t exist. So, to capture a lot of that, they have up classified (changes to the rules for a device classification, see above).This is especially true for electrical hardware, firmware, software, cybersecurity, and the mobile applications.The rules and expectations for these areas have gotten more explicit and stricter.
It seems like a big part of MDR is that there’s going to be more traceability needed, unique identifiers, and more labeling for each device. Do you see that impacting the way organizations develop medical devices? And what’s the best way to capture that information, or anything else related to post-market surveillance?
Satyajit Ketkar:
So, for the UDI implant card, etc., that’s new, and wasn’t there before. So, everyone’s starting from scratch.
Some people were kind of doing UDI (Unique Identification Number) labeling in the Instructions for Use (IFU) or maybe the eIFU, but it wasn’t to this level of rigor. You must now maintain vigilant records for all of this, for the entire life of the product including part of the post-market. There’s supposed to be a whole database that they’re putting together to keep track of all the vigilant reporting and the post-market reporting, that’s called EUDAMED. There are now updates to the Med Dev guidance(s) on clinical and post-market that are going to be referred to within the regulation. So, I would recommend that manufacturers continue to follow and comply to that. Beyond that, it’s pretty much the same thing.
What’s changed significantly is the clinical follow-up. There’s a whole new guidance on clinical safety and performance that they must maintain, which is part of this UDI tracking and vigilant tracking. Also, there is a periodic clinical evaluationthat mustbe conducted. There’s aperiodic safety and performance report as well that needs to be updated thatmust be included in the vigilance and post-market surveillance. These, and the UDI, are all new with MDR. Before, you would do this once in your three or five-year review cycle, and then that was it. Now, you must basically maintain these all the time as part of your post-market surveillance.
This doesn’t introduce a development burden, but it is a lifecycle burden. You must maintain these throughout the life of the product, which could be 15, 20 years, especially if it’s an active implantable, a pacemaker, or any sort of a therapy device.
One of the other things that I would recommend on the regulatory side or the system side is to review Annex 1, which contains the safety and performance requirements called GSPR (General Safety and Performance Requirements). The MDD has a set of 13 EssentialRequirements (ERs), and the AIMDD has16. The MDR has 23 GSPRs. This might seem less overall but each one of these requirements has 10 to 30 sub-sections that you must adhere to. So, in reality, the requirements have more than doubled.
Typically, what happens is that when somebody in a regulatory group or clinical group puts together their submission package, they’ll review all these essential performance and safety requirements to make sure that they’ve got all the documentation or all the deliverables. They’ve gotten very explicit as to what you need to provide. One example, specifically Section 17.2, which talks about electronic programmable systems and software. They’ve gotten specific about cybersecurity. They’re catching up with the FDA on cybersecurity and software lifecycle. That’s just one example, but there’s a whole bunch of other ones in there. These are new. So, that is something that they need to be aware of as they do their preliminary risk assessment and overall risk managementto comply with ISO 14971.These will be additional inputs for decomposing their system requirements. They need to be aware of the new and existing requirements.
Learn how Jama Software can help medical device developers better manage risk with ISO 14971 by downloading our white paper.
Jama Software:
Do you think MDR will have an impact on the way people create and approve requirements? Do you have any thoughts on how the requirements managementprocess might change?
Satyajit Ketkar:
I’m moderately confident that when it comes down to a very specific phase(s) of the development lifecycle, where you’re decomposing requirements and creating your traceability, from design, implementation, through to verification, that it should stay the same. It’s just the details of what you’d have to maintain that are going to change. The process should stay the same. Because remember, there are two parts of what you do: You create a DHF to maintain for yourself as a manufacturer, and then there’s a subset of that that you send to a notified body. The burden of what you must maintain for your own records goes up quite a bit. The process stays the same. It’s just what you must keep track of now that has gone up. The traceability is going to go up a little bit, but the process of decomposing requirements to subsystem, technical, and doing the architecture design, etc., all the current and standard processes should stay the same.
Jama Software:
Do you see this impacting the way global companies communicate or collaborate as part of MDR?
Satyajit Ketkar:
Yes: it’s going to require quite a bit more communication. You must stay aligned a lot more closely because there’s a lot more burden. There’s a lot more meat on the bone you must provide. So, if you have a distributed team across multiple regions, you need a central repository to maintain all these documents.
Jama Software:
You mentioned the DHF and that burden of proof is going to really be on the company.Does it impact the way folks adhere to ISO 13485 or Quality Management Standards at all?
Satyajit Ketkar:
No, it should not. Since it is a brand-new regulation, one of the things they’ve done with the MDR is adopt all the latest harmonized standards. That includes the latest QMS standard, latest risk management, etc. So, if you’re up to date on those, then you should be fine. You should not need to change your QMS to adhere to the MDR as long as you are maintaining the state-of–the–art.
Jama Software:
You mentioned doing earlier risk management followingISO 14971. Are there any impactsto the risk management process?
Satyajit Ketkar:
No. It’s kind of the other way around. MDR has taken account for the updates to all these other standards, especially the updates to risk and QMS. So, if you follow the MDR and you’re up to date on your revision of these other standards, you should be fine.
Jama Software: Is there anything else that you think we missed, or you think should be top-of-mind related to MDR?
Satyajit Ketkar:
I want to be very specific about what I just said earlier in following the harmonized standards and maintaining the state-of-the-art. If you’re not up to date on the latest standards, for example, if you’re not following ISO 14971:2012, or if you’re not following QMS:2016, if you are behind and you want to jump up to the MDR, you’re going to have a pretty heavy lift because you must comply to the latest revisions as well. It’s all included in the harmonized standards list now as this list has been updated. So, if you’re already doing it (not just complying to the current harmonized standard but aware of the state-of-art), then you’re fine. If you’re not, there’s going to be some uphill climbing that may be required.
What’s typical in my experience is that people try to keep up with the standards more often than the regulations. But they’ll see a big jump in the regulation, and they don’t want to jump straight into the regulation and find out that they’re still back in 2006, they haven’t revved up to the latest one (typically the state-of-the-art), and they’re stuck. So, just want to make sure that manufacturers are aware of that.
Jama Software: How do you think a dedicated requirements management platform like Jama Software fits into all of this?
Satyajit Ketkar:
If you’re already using Jama Software, you won’t have to change the process too much. The only thing that this new regulation is going to change is that people are going to want to use Jama Software more because it’s going to become necessary. There’s just too much from a QMS perspective and a development process perspective to try to do manually.
Because of this new regulation, it’s going to slow down their development lifecycle at first, because they are now learning about this new regulation. On top of that, if you’re doing things manually, if you’re using Excel spreadsheets or Word documents, whatever it may be… You might have a very simple project, but now you must add that manual process burden on top of this new regulation. So, it would reduce that process burden greatly if you had a solution like Jama Connect that would handle a lot of this for you. If you could just enter in all your requirements into a lifecycle management tool that then allows you to create trace tables, allows you to create upward, downward decomposition, and presents that in a very clean way where you can export it to a DHF artifact. That saves time and effort.
That’s the whole point, is to save time, because you already are in a process and a regulation change.Tools like Jama Connect will accelerate that because I do foresee a major slowdown in the lifecycle of medical devices with this new regulation… especially for startups. They’re going to have a tough time picking this up. So, to have a platform like Jama Connect out there to aid in this is going to be very important.
Jama Software: What resources are available to people that you know of to help them comply with the new standards?
Satyajit Ketkar:
Most of the approved, certified, notified bodies in Europe — some of the ones that I mentioned before like BSI, TÜV SÜD, DEKRA — are a great resource. They should have a quick cheat sheet on their website. You can also go online to any of the medical device journals, and they have a pretty good high-level breakdown of EU MDR. It’s more on the regulatory side than a development-process side, but it gives you the breakdown of what articles are necessary, what annexes need to be reviewed, and what order to do it in.
Medical technology companies, especially those developing medical devices with increasing complexity and product variations, are faced with a wide range of methods for managing risk. The toughest part is often deciding which specific methods and data points need to be captured to prove that all angles of risk analysis for product development took place. Against this backdrop of constant innovation, medical device manufacturers face unique challenges to comply with significant regulatory changes in Europe and the United States — some that could support new product development, while others could hinder it.
Changes in compliance standards, such as the new Medical Device Regulation (MDR) in Europe, include sweeping changes regarding how companies and distributors demonstrate the safety and efficiency of products. Companies are required to show clinical evidence for all products as well as provide systems and documents to remain compliant during the device lifecycle. One-way companies are reacting is by adopting modern requirement management solutions for improved agility and flexibility. With the ability to trace all aspects managed in one place, unified cloud requirement management applications can help maintain compliance and facilitate engaged collaboration across geographies. Evolving a regulatory environment means it is no longer feasible to have unrelated content without convenient access.
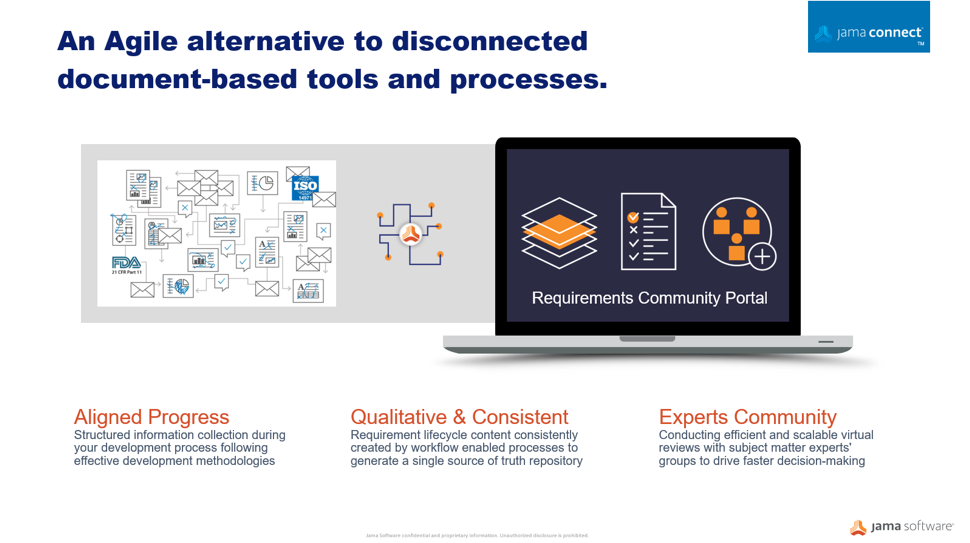
Whether regulatory authorities are tightening rules around medical devices or responding to market innovation, manufacturers must be prepared to demonstrate their products are safe and effective across the total lifecycle using a more comprehensive array of data sources. Critical factors for medical device makers must transform the way they innovate, design for manufacturability, and differentiate their products in the market. A centralized Agile alternative to disconnected, document-based processes enables pathways to faster product development, improved quality, and compliance to safety regulations.
How can you modernize and where should you start to meet these compliance challenges while streamlining your development processes and creating efficiency? Three improvement strategies can be used to insert risk control as a crucial part of the medical device development framework to comply with ISO standards and EU Medical Device Regulation (MDR).
Three Strategies to Streamline Workflow and Strengthen Collaboration when Developing Medical Devices
With the magnitude and complexity of data expanding, medical device firms are working to assemble and analyze data faster. Much of this data is often spread out across information silos. As a result, medical device companies are increasingly looking to modern clinical requirements and safety management systems, including central data capture, that collect and clean this clinical data, all in one manageable platform.
According to a study by the Emergo Group, nearly 60 percent of medical device builders say clinical data management rules are the most challenging component of Europe’s new MDR. An equivalent percentage of companies say they don’t have a strategy for compliance. Having an approach that incorporates a complete view of data can help with the preparation and ongoing compliance. To have a plan of action, three strategies that can help you to focus on a trustful quality and efficient control of developing advanced medical products.
Establish a Single Source of Truth Framework for Reliable Insights
Through medical device development, many disparate documents are gathered into a collected set of information and submitted to regulators. This auditing process can be a time consuming and very manual exercise often done too late in the development cycle. Optimizing workflows to centralize information sets in an item-based approach where processes and documentation move from the document-centric model to a relational database approach can protect your quality of building new products and services. A workflow-driven, traceable methodology, and managing all information sets in real-time brings better handling to safety-controlled development cycles, and maintains a continuous state of acceptance readiness.
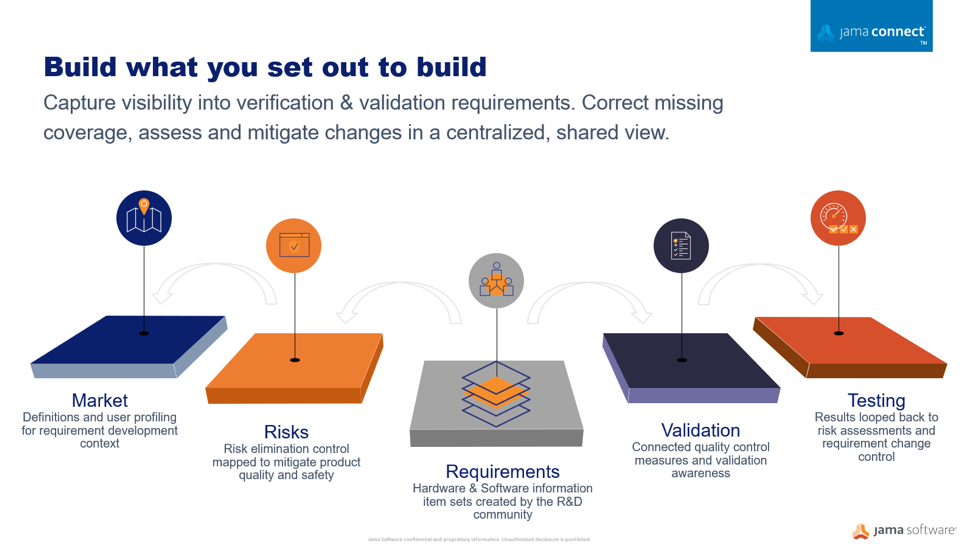
Streamline Risk Control with Requirement Definitions
Using a centralized information gathering approach, you can author the risk process directly connected to your development work. Your risk acceptability criteria can be defined and documented as part of the risk assessment related to the required development specifications. Moving to connected risk-requirements workflows, provides a collaborative place where the risk management plans can be viewed holistically by the entire development team in the context of the medical device development environment where requirements, specifications, validations, and risks are analyzed.
Enable Your Expert Community
Building complex products is never a one-person show. Medical device manufacturers when implementing tools, resources, and techniques to develop safe and successful products faster. A substantial factor for success is the team’s understanding and engagement at every aspect and stage of the development lifecycle, creating a robust traceable collaboration community and the ability to initiate a flexible evaluation process. These factors can have a significant impact on achieving a timely and cost-effective control that assures product quality due to a risk embedded impact analysis.
Medical device developers can optimize the development cycle by bringing risk control and development data together in an information-centric and automated fashion and instead focus on quality, innovation, and efficiency, as well as time to release, allowing medical device builders to recognize new product revenue within shorter release cycles.
Early transitions to renewed regulations, such as the new European MDR/FDA standards, are critical in giving products a competitive advantage. Companies can turn this around into an opportunity by adhering to the following recommended steps:
Implement a strategic, information centric approach early on to ensure market compliance and build a trustful reliable data source
Handle your risk control elements directly connected to your traced development activities
Make use of Jama’s consulting expertise and the available medical assets such as ISO 14971, FMEA, FTA, and 21 CFR Part 820 to rollout your renewed strategy based on industry best practices.
To see more information specific to the medical device development industry, we’ve compiled a handy list of valuable resources for you!
 Mean for You") In this post, we pull out key takeaways from a recent whitepaper written in conjunction with Beanstock Ventures on the new EU medical device regulations (EU MDR) and how they might impact medical device development.
In this post, we pull out key takeaways from a recent whitepaper written in conjunction with Beanstock Ventures on the new EU medical device regulations (EU MDR) and how they might impact medical device development.