In many ways, 2021 was a continuation of the changes brought about in 2020, a year that’s been described as “unprecedented” and “unparalleled.” In a unique way, 2021 has offered us an idea of evolving innovations and technology on the horizon for teams across industries. These changing conditions will present a variety of new landscapes and will offer unique challenges, opportunities, and more than likely, many surprises.
As we enter a new year of further changes, Jama Software asked select thought leaders – both internal and external – across various industries for the trends and events they foresee unfolding over the next year and beyond.
In the third part of our five-part series, we ask Adrian Rolufs, Director of Solutions Architecture from Jama Software, to weigh in on product and systems development trends he’s anticipating for automotive development in 2022.
Read our other 2022 Industry Predictions here:Part One – Engineering Predictions, Part Two– Medical Device Predictions, Part Four – Aerospace & Defense Predictions, and Part Five – Insurance Development Market Predictions.
Q: What product, systems, and software development trends are you expecting to take shape in 2022?
Adrian Rolufs, Jama Software:
2022 will continue much like 2021. Many established automotive companies are in the process of modernizing their development processes and tool chains. These companies are looking to adopt Agile principles to allow them to execute faster and adopt modern tools that better support their new process. Many of the startups established in the last couple of years are maturing and discovering a need to add more robust processes to ensure that as they bring products to market, they maintain compliance with the safety and quality standards in automotive.
Due to the global chip shortages in 2021 that had a huge impact on automotive OEMs, we’ll continue to see a focus on ensuring that there is a sufficient supply of automotive grade chips.
Q: In terms of product and systems development, what do you think will remain the same over the next decade? What will change?
Adrian Rolufs, Jama Software:
Over the next decade, I expect that automotive systems development will continue to place an emphasis on software defined features. OEMs will continue to heavily invest in their software development capabilities and an ongoing focus will be placed on quickly delivering new software features while maintaining quality.
A major change that I see coming is a wider adoption of vehicle variation through software differences rather than hardware differences. Tesla has already led with this approach, but I expect to see more manufacturers ship vehicles with a minimum variety of hardware, and options provided through software configuration instead.
Q: How do you foresee regulations shifting in Automotive Product and Systems Development over the next decade?
Adrian Rolufs, Jama Software:
The current big shifts will continue over the next few years. An increased focus on cybersecurity is already happening and will be a major factor for automotive companies to adapt to over the next few years. With over-the-air updates quickly becoming a mainstream feature of new vehicles, a huge focus must be placed on ensuring safety and regulatory compliance as updates are rolled out.
Autonomy is an area where new standards have been recently developed, like UL 4600. I expect to see significantly more regulations around autonomy in the next decade to create a framework for bringing fully autonomous vehicles to market.
Q: Any major disruptions to Automotive Product and Systems Development industry you’re anticipating in 2022?
Adrian Rolufs, Jama Software:
New electric vehicle manufacturers like Rivian and Lucid are starting initial production now and are planning to ramp up production in 2022. If they are successful, this will put additional pressure on established OEMs to execute on their own electric vehicle programs even faster than they already are. This will likely have a cascading effect felt across the industry.
Q: What sorts of process adjustments do you think development teams will need to make to be successful in 2022?
Adrian Rolufs, Jama Software:
With the new focus on allowing for remote work, the traditional dependency on tribal knowledge and the heroic efforts of individuals will not be enough for companies to be successful. Product development knowledge has to be captured in systems and kept up to date so that remote workers can still be productive. This will continue to push for more modern tooling and increased enforcement that is used correctly. Capturing accurate requirements, establishing traceability, and being able to keep track of it all in a highly iterative fashion will be critical to ensuring success.
Q: What do you think will be some of the differentiators between a company surviving to see 2030, and those that do not?
Adrian Rolufs, Jama Software:
The established companies that survive to see 2030 will be those that adopt modern development practices fast enough to stay competitive and continue to stay relevant in the market.
For the startups, the biggest challenge is maintaining strong enough financial backing to make it to mass production of their product. Many of the existing startups will be acquired or closed before their products ever make it to the market. Those that succeed will have balanced the needs of fast time to market with robust product development processes that ensure quality.
Q: Where do you see Jama Software fitting in as the product development landscape evolves, and what can our customers expect as 2022 approaches?
Adrian Rolufs, Jama Software:
Jama Software will continue to provide the most useable requirements management, test management, and traceability solution on the market. Jama Software will provide solutions to the companies that are striking a good balance between quality and fast execution.
Thanks for tuning into our 2022 Predictions Series! To see some of the incredible products, software, and systems our customers are building with Jama Connect, visit our customer stories page.
The Top Challenges of Property & Casualty Insurance Product Development
Today’s P&C Insurance landscape is both vibrant and facing major disruptive change. With the top 10 carriers commanding well over 70% of the personal lines, auto and home insurance market and hundreds of additional carriers making up the balance, things were already highly competitive. Growth in digital channels has altered insurance shopping and buying behaviors adding to the commoditization trend of insurance products, making it easier than ever to switch carriers, especially for auto insurance. Telematics for auto, IoT, and smart home technologies are engaging consumers differently and offer the benefit of avoiding and mitigating losses as the industry shifts towards risk prevention from traditional sole protection and indemnification models. These are just a few noteworthy trends among a host of automation efforts, new insurance models and products – all happening throughout the recent insurtech movement.
Commercial lines continue to become more vertically specialized, particularly in small business lines. New insurtech entrants tend to focus on disrupting select product lines where larger, multiline established carriers may underserve certain business owners and markets.
The P&C industry is moving through a broad range of innovation with over $15 billion during 2021 in venture capital investment and scores of outside influences converging on this 100+ year industry like never before. Meanwhile, established carriers are pushing through multi-year legacy system transformations, heavily investing in innovation while striving to modernize core processes which have historically been manual and people centric.
Each of these variables are pressuring insurers to lower operating costs while automating and launching new products to differentiate and compete in protecting market share with only a handful of carriers capable of gaining share. New products and revisions to existing insurance products demand calls for rapid speed to market vs. traditional launch times that typically span up to a year or longer. The insurance product development process is highly dependent on effective requirements management and constant cross-functional team collaboration, which is vital to avoid costly delays, defects, and premium refunding due to undetected errors.
Insurers are highly people driven organizations and are well-known for having siloed organizations given the pervasive scale and specialized work teams. When it comes to developing and launching a new insurance product – it truly takes a village. Stakeholders and contributors span from several highly specialized areas, including actuarial, pricing, underwriting, legal, claims, marketing and distribution to name a few. Thus, collaboration does not always come naturally nor easily. Associate engagement and company culture are top priorities and having the right tools and technology can make a big difference given that much of insurance product development relies on, legal research, specialized rate making expertise, approvals and decisions often made through emails and organized via spreadsheet tracking. Some use collaboration platforms and shared drives, which can be helpful but lack versioning, baselining, uniformity and most importantly change and impact analysis due to ineffective live traceability.
While carriers are adopting automation, much of the attention is on gaining new customers, services, distribution, and overall efficiency gain. Digitization, self-services, and artificial intelligence for predictive analytics are a few examples. New data sources, such as geospatial and personalized data and image analytics are being leveraged from underwriting to claims management. Automation of internal processes such as pricing (ratemaking) and product development have seen lesser insurtech funding and focus and tend to be less obvious or viewed as more complicated areas to solve in comparison. Leading insurance companies have recognized these processes must receive greater attention to drive competitiveness and increased speed to market.
Speed to market and quickness to revise and modify products have many obvious benefits in today’s highly competitive P&C insurance marketplace. Most would agree that the typical insurance policy itself is quite complicated. A look behind the scenes at the process of pricing, rate making and filings with respective state departments of insurance along with attendant policy administration and IT programming is where the real complications begin. After all, insurance premiums are priced on an individualized basis by design. In other words, each consumer, or segment of customers more realistically, have differing risk profiles and thus different price points which can number in the thousands of different rates for a single product.
The US insurance industry is regulated on an individual state level through each respective department of insurance, responsible for approving products and regulating rates among other consumer protection focal areas. Additionally, each state has a bevy of insurance regulations, statutes and common laws and each insurer has differing marketing, regional considerations and so much more to balance when it comes to launching new products. With all of this in mind, it is easy to see how errors can be made or interpretations might differ, only to be discovered at a later time, which adds cost and increases regulatory scrutiny.
There is a lot to be said about doing things right the first time by being able to test, trace and apply impact analysis early and often. The consequences of product defects are most visible during so-called market conduct studies performed by departments of insurance which can result in penalties, fines, and premium refunds. Or, a carrier may self-discover a defect and proactively take measures. Either way, the amount of resources and efforts to identify root causes and deploy remediation measures are extremely costly and often harmful to the brand itself. Less visible scenarios can occur during product development cycles and simply result in time delays, re-work and fixes which account for the extra time to launch or altogether delay and avoid making revisions to existing products. The stakes are high in terms of costs and missed opportunities. Insurers investing in tools and technologies to build insurance products better and more efficiently can enjoy a competitive advantage.
In many ways 2021 was a continuation of the changes brought about in 2020, a year that’s been described as “unprecedented” and “unparalleled.” In a unique way, 2021 has offered us an idea of evolving innovations and technology on the horizon for teams across industries. These changing conditions will present a variety of new landscapes and will offer unique challenges, opportunities, and more than likely, many surprises.
As we enter a new year of further changes, Jama Software asked select thought leaders – both internal and external – across various industries for the trends and events they foresee unfolding over the next year and beyond.
In the second part of our five-part series, we ask Steven Meadows, Solutions Lead from Jama Software, and Ryan Moore and Carleda Wade, Solution Consultants from Jama Software – along with Thierry Marchal, Program Director for Healthcare Solutions at Ansys, and Ivan Ma, Medical Device Program Leadership, to weigh in on product and systems development trends they’re anticipating for medical device teams in 2022.
Read our other 2022 Industry Predictions here: Part One – Engineering Predictions,Part Three – Automotive Predictions, Part Four – Aerospace & Defense Predictions, and Part Five – Insurance Development Market Predictions.
Medical Device Predictions
Q: What product, systems, and software development trends are you expecting to take shape in 2022?
Steven Meadows, Jama Software:
Artificial Intelligence (AI)
AI, and in particular machine learning (a subset of AI), has been expanding rapidly over the past decade and now has a market of over $6.7 billion across med tech. I see this trend continuing with extra guidance from the FDA helping developers produce safer software with AI elements. AI is particularly exciting as it can enable streamlined MRI and CT scans, instant blood and at-home rapid testing, and a whole host of otherwise manual and often error prone activities.
Minimally invasive devices
Another trend I see in medical continuing in 2022 and beyond is the rapid growth and development of minimally invasive devices. Open heart surgeries are becoming a thing of the past, with cardiothoracic applications becoming the norm. Minimally invasive devices are also becoming heavily utilized throughout most orthopedic surgeries, and increasingly in urology procedures. We have seen a lot of start-ups attempting to bring the next best minimally invasive device to market.
Wearables
Medical wearable devices demand continues to rise, with apps on wearable devices beginning to provide recommendations to users, to improve their health. Heart rate and temperature sensing continue to be the most popular features on wearable tech today but there is increased investment in smart glasses, ‘earables’, and clothing.
Software development
Software will continue to be an integral part of many medical devices next year and beyond. We consistently see that our software customers have adopted, or are transitioning to, an Agile methodology when creating their medical system, a shift that has been happening for some time. Gone are the days of slow waterfall-based development practices.
As I mentioned before, AI based software development will continue to trend and grow next year, and AI software development practices will be geared around a tailored regulatory framework, good machine learning practices, and a patient centered approach. The FDA will continue to refine guidance around AI/Machine Learning (ML.)
Cybersecurity and in particular data security will continue to be a top priority for our software customers, with data breaches and hacks on the rise. The FDA is planning to soon release extra guidance around cybersecurity in medical devices with a particular emphasis on quality system considerations and content of premarket submissions.
Ryan Moore, Jama Software:
I anticipate a trend toward more software-basedtoolsets and digitization. Also, an increased amount of automation and robotics.
Carleda Wade, Jama Software:
I expect greater integration of software tools such as with Requirements Management software with eQMS software.
Thierry Marchal, Ansys:
The dramatic COVID pandemic has amplified a trend that appeared a decade ago – progressively calling for the adoption and deployment of in silico medicine. Stormed with the COVID pandemic, the world could not wait for 10 to 15 years to get a new vaccine fully tested and approved using a traditional approach. Furthermore, this COVID disease is impacting people differently, while elderly people are impacted the most, possibly leading to long term disabilities and treatments.
EDITOR’S NOTE: According to The University of Sheffield’s Insigneo Institute, In silico medicine (also known as ‘computational medicine’) indicates modeling and simulation technologies that directly contribute to the prevention, diagnosis, prognosis, treatment planning & execution, or management of the disease. In silico methods complement traditional in vivo approaches (working with animals and human beings) and in vitro testing (working in a lab.)
Thierry Marchal, Ansys:
This pandemic highlights what we have been observing for decades: with an aging population, the cost of chronic diseases (especially for old people) is weighing a lot on the healthcare cost. The solution will come from personalized medicine and preventive medicine combining pathologies detection in its early stage to treat diseases before they impact people. The continuous monitoring of patients to detect pathologies early are calling for e-health and mobile health (m-health): wearable and implantable devices continuously and safely watching the patient and soon feeding your own digital twin or Personal Digital Avatar (a computer model of yourself, connected with you, and properly stored in the cloud to guarantee your data privacy) is now an emerging trend. Digital twins will be a crucial innovation for the software necessary to use patient specific data to predict the evolution of the connected patients.
As medical innovation will be essential soon, this evolution cannot be slowed down by an extremely long and costly regulatory approval process: a digitalization of drug and medical device approval, including in silico (clinical) trial is another major trend that we are observing.
Ivan Ma, Medical Device Program Lead:
Technologies that enable capabilities via telemedicine with more than just a face and a voice over the screen will make “seeing” the doctor digitally as common place as working from home.
Protecting patients, physicians, and staff by reducing or eliminating exposure to harmful forms of imaging such as fluoroscopy will always be a valuable endeavor.
Q: In terms of product and systems development, what do you think will remain the same over the next decade? What will change?
Steven Meadows, Jama Software:
A lot of the areas I mentioned that will trend in 2022, will more than likely trend over the next decade.
AI, not only in medical but across most industries, is on the rise and patients will continue to see improved outcomes, quicker diagnoses, and a better quality of life.
Medical wearable technology will continue to evolve, with improved functionality helping keep us fitter and safer.
Minimally invasive devices will continue to be expanded across different medical areas, improving recovery times, and surgery outcomes. Increasing emphasis on cybersecurity will resume, to prevent malicious actors from hacking sensitive data and for connected systems to remain operational.
COVID-19 is not going away any time soon so a reliance on collaborative product development tools, like Jama Software, will continue to be an integral part of any organization placing an importance on quality and overall product and patient outcomes.
Ryan Moore, Jama Software:
Medical devices will still be comprised of mainly hardware (HW)/software (SW), while automation and robotics will be a driving force.And users will still need to power devices.
Carleda Wade, Jama Software:
Medical device development will mostly stay the same as the regulations are fairly stable.However, I do expect increased usage of software in the process since much of the workforce may now be working in various locations. I also expect to see an increase in AI and Software as a Medical Device (SaMD)as a whole in the industry.
Q: What are some of the biggest challenges you think engineering firms will be working to overcome in 2022?
Thierry Marchal, Ansys:
Contrary to other industries, innovation in healthcare faces the challenge of human variability: as we are all different, it is not acceptable that a treatment would work well for a few people and poorly for most others. Using computer modeling and simulation (CM&S a.k.a. in silico methods) is a cost-effective way to test new treatments efficiently without compromising with patient safety, on large cohorts of virtual patients.
An accurate prediction of the treatment outcome for a given patient will require combining traditional modeling techniques with biological models, often extracted from big data observations using AI.
Finally, the community remains skeptical about the reliability of computer model and digital evidence to predict correctly and accurately what is happening in the real life. This credibility challenge will be continuously addressed by more validations and comparisons with in vitro and in vivo data.
Ivan Ma, Experienced Medical Device Developer:
The development of medical device hardware will always require teams to gather in front of and handle hardware. As the pandemic continues, and in-person work still not back to what it once was, it is time to think of strategies that maximize time on developing hardware with minimal people in the room. Could digital Operating Room technologies be reconfigured and brought in to support the verification and validation phases of medical device development? The tenets are the same. Minimize the number of people in the room, allow the expert to drive the hardware from afar, make data acquisition, observation and learning access as easy as clicking on a link.
Q: How do you foresee regulations shifting in medical device product and systems development over the next decade?
Steven Meadows, Jama Software:
Although AI has been utilized in medical device and life science products for decades, guidance has been lagging. It’s clear to see that AI has incredible benefits for patients, and so there will continue to be increased regulatory guidance available, to help developers build products which contain AI in a safer and standardized manner. Check out a blog Jama Software authored around machine learning in SaMD and shifting regulations here.
The medical device regulation (MDR), which was accepted and implemented in the EU in 2017, has been amended over the past few years. The latest change focuses on increasing the responsibility and accountability of medical device companies throughout the entire development of a product. Expect more guidance over the next decade, as gaps are addressed to ensure product safety is at the forefront of any market clearance.
Ryan Moore, Jama Software:
I would expect compliance and regulations to be more defined as we move forward. Currently, there is a lot of grey area in how standards/regulations are interpreted. The FDA will need clearer guidance for modern technology that arises
Carleda Wade, Jama Software:
With an uptick in AI and SaMD I expect the FDA and foreign regulatory bodies to begin providing more clarity on how these complex systems and softwareneeds to be developed, validated, and maintained.
Q: What changing regulatory guidelines do you anticipate having an impact on companies in 2022?
Thierry Marchal, Ansys:
The US authorities developed, without any doubt, the most advanced regulation in terms of adoption of computer model results for the regulatory approval process. However, the number of published cases reporting the actual use of in silico methods by sponsors remains limited. New results will be published in 2022 further encouraging companies to confidently follow this process.
The European Medical Device Regulation (MDR), in application in Europe since May 2021, is opening the door to digital evidence and in silico approach; unfortunately, the process to validate a model and report simulation results has not been clarified yet. Similarly, as the European authorities are updating their pharmaceutical strategy in 2022, this document is expected to make references to computer modeling and simulation.
In the rest of the world, we observe a growing interest for in silico methods and the need to regulate this approach. After years of compiling information and experience from other parts of the world, we are expecting that some Asian authorities will start to communicate about this topic in 2022.
Q: What sorts of process adjustments do you think medical device development teams will need to make to be successful in 2022?
Steven Meadows, Jama Software:
One of the biggest process issues we see across our customers is that they view quality as a secondary function and a necessary checkbox activity once a product is developed, or close to being finalized. Because of that, products tend to contain more defects, resulting in more in field CAPAs, negative patient outcomes and even have a greater chance of being recalled. Development teams should ensure quality is prioritized from the get-go.
Although we have noticed a large shift with software teams adopting an Agile approach when it comes to development, we can’t emphasize enough the importance of adopting a lean and iterative approach.
Ryan Moore, Jama Software:
Continue to follow standardized process with focus on aligning with FDA / compliance in parallel with building safe products
Carleda Wade, Jama Software:
Teams will need to find the sweet spot of trying to collaborate and innovate while not being physically in the same location at the same time. Due to the pandemic, many teams will now transition to being fully remote permanently or have a hybrid schedule and it may make things more difficult when developing new products with a cross-functional team.
Q: From an engineering toolset perspective, what are some of the processes you believe forward-thinking firms will be working to leverage or incorporate into their process and why?
Theirry Marchal, Ansys:
As we see the healthcare community adopting in silico method with more enthusiasm both for design and regulatory approval (not mentioning emerging clinical applications), the software community is rushing to deliver the supporting in silico tools. We could mention three major avenues followed by Ansys.
As in silico clinical trials requires a large number of simulations, specific Simulation Process and Data Management (SPDM) tools structuring the digital evidence following the ASME VVUQ40 standard will greatly facilitate the adoption of this approach. Ansys is customizing its Minerva tool in a Minerva VVUQ 40 template for medical device and pharmaceutical companies.
It is crucial to model the behavior of a new treatment in its working environment, usually the human body. Human organs such as the heart, the lungs, and the brain are extremely complex to model and validate: this requires a collaboration of various actors. Ansys is closely working with leading academics, global healthcare companies and startups, and clinicians to continuously update and validate virtual hearts, lungs, and brains in collaboration with a large ecosystem.
Although everybody recognizes the potential of digital twin and Personal Digital Avatar, the necessity of developing quasi-instantaneous modeling capabilities despite the complexity of the model is a real challenge. Ansys is taking advantage of its experience with other industries already using digital twin of equipment to help the pharmaceutical industry to develop digital twin of drug manufacturing equipment and initiate the first steps towards Personal Digital Avatars.
Ivan Ma, Medical Device Program Lead:
Engineers need tools that minimize the volatility and error that occurs when bringing hardware and electronics from the drawing board to parts in hand. How do organizations reduce hardware prototype cycles, recognizing that even if the design is right on paper, the production of that design can pass through many hands before it is made into a real part, thus decreasing the probability of a successful build.
Q: What do you think will be some of the differentiators between a company surviving to see 2030, and those that do not?
Steven Meadows, Jama Software:
Along the lines of what I mentioned companies should focus on in 2022 to be successful, medical device companies that embrace a proactive approach to quality(Link here – https://www.jamasoftware.com/blog/three-ways-to-proactively-vs-reactively-incorporate-design-controls-in-medical-device-product-development/ )will ultimately find fewer issues with their products, improve customer satisfaction, and stay competitive for the foreseeable future. Companies that invest in best-of-breed medical device development solutions like Jama Connect will have the upper hand in reducing risk and complying with various standards and regulations.
New cutting-edge technologies in the medical field – like robotics nanotechnology, AI, and wearable health tech devices – bring complexities for medical device companies and risk for patients and consumers. Jama Software will continue to serve as the leading solution and partner to help innovative companies bring medical products to market, in a collaborative, safe, and efficient way.
Defined process, building quality products that meet a specific market need
Carleda Wade, Jama Software:
Truly understanding the market needs and being willing to allocate resources so that you become a pioneer in a certain industry will help a lot of companies to succeed; however, having passionate and qualified personnel will be the biggest success factor. Companies that only want to develop “me too” devices will struggle to gain market share, and even if they are able to survive it will be difficult for them to thrive.
Q: Where do you see Jama Software fitting in as the product development landscape evolves, and what can our customers expect as 2022 approaches?
Steven Meadows, Jama Software:
Jama Software provides incredible value for hundreds of medical device companies ranging from start-ups to established corporations. With COVID-19, we’ve seen an uptick in customers looking for an easy-to-use product development tool that offers differentiating collaborative capabilities to keep people connected throughout different design stages. Jama Software continues to invest heavily in the application, and we will see improved core product capabilities as well as integrations, strengthening the tool’s ability to seamlessly work alongside other systems.
Ryan Moore, Jama Software:
Jama Connect can be used as the core toolset for requirements, testing, and review/approvals. Jama Software’s ability to branch out into further workstreams (in order of priority: risk management, test automation, modeling/design outputs) will bring exponential value to teams using Jama Connect.
Carleda Wade, Jama Software:
I can see Jama Software bringing value to not only the start-up but the established company in the future. As a medical device company, the first goal of the company is to make a good product that meets a market need, this happens even before establishing a quality management system in many cases. Having a tool like Jama Connect will ensure that as products are developed that all the bases are covered. Later, when it is time for regulatory approval having a tool like Jama Connect in place will make the regulatory submission process very simple.
Thanks for tuning into our 2022 Predictions Series! To see some of the incredible products, software, and systems our customers are building with Jama Connect, visit our CUSTOMER STORIES PAGE.
This is Part 2 of a series examining the role legacy requirements management solutions, such as IBM® DOORS®, play in introducing project risk to the product development process. To read Part 1, visit Why Migrate, Why Now?: Part 1
5 Common Migration Myths Debunked
Transitioning to a new solution doesn’t have to be challenging; however, there are some assumptions that mislead us into thinking that difficulty is inevitable. Consider the following myths:
MYTH 1: Migrating away from IBM solutions will be more expensive. The amount of work that goes into upgrading to IBM DOORS Next or transitioning to a new RM solution is the same, differentiated only by the quality of the tool and services available to help with migration. An option other than the DOORS family is most often a better fit for your organization.
MYTH 2: Customization will carry over to DOORS Next. You spent a lot of time customizing IBM DOORS and may believe those customizations will transition seamlessly to DOORS Next. However, this isn’t the case and is the reason selecting a different solution doesn’t involve more work.
MYTH 3: DOORS is already deployed and cheap to maintain. Continuing the current path with IBM DOORS is an expensive option in the long term, and often requires dedicated personnel. Switching to an alternative RM solution can improve efficiency while saving money.
MYTH 4: Business disruption is too difficult. The right RM tool will empower teams to effectively hit deadlines, collaborate, and improve business outcomes.
MYTH 5: The user experience will suffer. Many people refuse to use DOORS due to a challenging user experience. DOORS Next is a completely new tool with a new user experience. Adopting a user-friendly solution allows teams to collaborate far more effectively as team members can accelerate concepts, designs, and validations for faster times to market.
The fact is, IBM® DOORS® is extremely outdated, and at some point, updates and support will inevitably end.
If you’re currently using DOORS, you likely know that moving to a different solution is necessary, and you might be considering DOORS Next. However, fast-shifting market dynamics require a new approach to accelerate innovation. As a modern alternative to traditional legacy platforms, Jama Connect® enables digital transformation with a more efficient and user-friendly approach to managing risk and compliance.
Customers agree, naming Jama Connect the overall leader (#1) in requirements management software on G2, outranking IBM DOORS Next for implementation time, adoption, ROI, and market presence.
Jama Connect is a proven IBM DOORS alternative with flexible and reliable solutions, including:
Operation in an IBM DOORS supply chain.“Innovative companies leverage Jama Connect to get up and running fast with a modern requirements management process that tightly aligns with industry standards and practices that support regulatory compliance. Organizations can connect to customers and suppliers that use IBM DOORS through Data Exchange for Jama Connect.”
Integration services. Jama Connect provides integration with key product development lifecycle tools.
Coexists with IBM DOORS. IBM DOORS is embedded in many organizations and may take some time to migrate completely. Progressive teams and divisions can get started on Jama Connect quickly while the larger organization works toward replacing existing programs over time. This approach is supported through a mix of integration, migration, and exchange services.
“Jama Connect lowers the complexity and burden of having to manually keep requirements, architecture and specifications all in sync and traced to each other. It’s a formidable problem that is virtually eliminated courtesy of Jama without the hassle of having to learn a clunky UI (IBM DOORS).” Alan M., Chief Product Officer
Jama Connect® created Live Traceability™ management which reduces project development risk by forming a digital thread through siloed development, test, and risk activities. The flexible platform is designed to support the end-to-end product development process with:
A simple, single repository so it’s easy for remote teams to gather, review and execute on requirements.
Structured reviews and collaboration — teams can elicit feedback, review product features in real-time with stakeholders and track critical decisions across teams and locations.
Change management throughout product development — end-to-end traceability and real-time collaboration improve visibility and make it easier to adapt to changes and track their impact.
Integrations across the ALM-PLM ecosystem.
Moving from IBM® DOORS® Next to Jama Connect® for Requirements Management
To adapt to shifting and ever-increasing challenges and complexities and keep pace with the competition, innovative organizations are now requiring best-in-class software to scale development, reduce risk, save time, and ensure compliance to quality and safety regulations.
Download this paper to learn how Jama Connect stands out above DOORS Next in the G2 Summer 2021 report for requirements management in the following areas:
Products, systems, and software development are only getting more complex and not modernizing your requirements management process will increase the probability of negative outcomes in your product development process.
As your team requires the ability to adapt, innovate, and grow, continuing to use IBM DOORS will become more difficult and will introduce significantly more risk in your product development process. Think about DOORS as a landline rotary phone that stopped being on the receiving end of upgrades after the industry switched to marketing push-button touchtone phones. (And now there are smartphones available, which are mobile and make you even more connected and productive.)
Transitioning to new technology provides your teams with the tools required to innovate, meet deadlines, and succeed. A modern requirements solution can help you to define, manage, and validate complex systems requirements while eliminating the risks and inefficiencies associated with documents and legacy systems.
Hear about the experiences of the more than 50 companies that have made the switch from IBM DOORS to Jama Connect.
Connect with us today to learn how you can enable end-to-end compliance, risk mitigation, and process improvement with our intuitive, award-winning requirements management platform.
Design history files were first required by the FDA in 1990 for medical devices. Learn how DHFs impact product teams, and get Jama Software’s DHF template.
In the competitive world of medical device development, medical device companies can’t afford to lag behind the pack. Streamlining the design and approval process is vital to getting products to market in a timely manner. Proper documentation is key to a smooth approval process.
One essential ingredient to medical device documentation is the Design History File, or DHF. While a bad DHF may not result in non-compliance, a good DHF can make the difference between a smooth compliance process and a bumpy one. To create a good DHF that streamlines compliance, design teams should know what to include—and what to avoid—as they go through the entire design and approval process.
What’s a Design History File, and How are They Used by Product Teams Working on Medical Devices?
Before reviewing all of the essential DHF ingredients, it can help to back up and establish what it is and why it’s important.
The Design History File is a collection of documents that describe the design and development activities of a medical device. Its purpose is to demonstrate that the device was developed using the design control process needed to meet FDA requirements.
Figure 1: Design Control Process for Meeting FDA Requirements
The DHF requirement was established as part of the Safe Medical Devices Act of 1990. The act describes the DHF in section 21 CFR 820.30, sub-section (j) as follows:
“Each manufacturer shall establish and maintain a DHF for each type of medical device. The DHF shall contain or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plan and the regulations of this part.”
There are two key terms in the DHF requirement: “establish” and “maintain.” “Establish” means that the development team must define, document, and implement requirements. Evidence is found in procedures, work instructions, drawings, and other similar documents. “Maintain” means that the team must review, approve, and update requirements. Evidence of maintenance is typically found in document control or change control systems of a company.
While a comprehensive DHF is vital to the FDA approval process, design teams will realize other benefits of maintaining the DHF, including:
Knowledge preservation and information sharing among key stakeholders.
What is the difference between DHF and DMR?
The DHF is one of three pillars of design compliance for medical devices:
Design controls: The moving parts of product design and development.
Design History File (DHF): The collection of records from design and development activities.
Device Master Record: The “recipe” for product manufacturing once the design is complete.
As one of the pillars of design compliance, the DHF is important, but it cannot hold up the design process on its own. It’s one leg of a tripod, and removing any one of the legs means that the entire design compliance process may tip over. Equally important to the process are design controls and the Device Master Record, or DMR.
What are design controls, and how does the DHF establish the design control process?
Design controls establish a plan to manage new product development; they are the process pieces that guide development along a predictable path to ensure that requirements are met. The Safe Medical Devices Act requires that manufacturers of Class II and Class III medical devices (and some Class I devices as well) implement design controls over the development process.
Design controls include:
Design inputs: The physical and performance requirements for the type of device. May also include types of materials needed, interfaces with other devices, and even packaging required, especially if the device must be sterilized.
Design outputs: Usually the final specifications for the device, typically documented in models, drawings, engineering analysis, etc. Outputs should trace directly back to initial requirements. Design outputs are the basis of the Design Medical Record (DMR).
Design review: A formal review by the design team and any other relevant parties, such as sales, marketing, manufacturing, etc. Documentation of the review should include the date, participants, version/revision reviewed, and results.
Design verification: This process verifies that the design output meets the requirements expressed by the design inputs and that specifications are correct. Documentation of this process must include the date, participants, version/revision reviewed, and the verification method and results.
Process validation: Process validation involves producing the device with a normal, low-level production process to confirm that the device will function as designed outside of the prototype process. Documentation of the process is required for the DHF.
Design validation: In this process, each manufacturer validates device design under specific conditions on small production lots or batches. The DHF documentation must include identification of the design, methods of production, date, and individuals performing the validation.
Design transfer: The process of transferring design into production, distribution, and installation specifications.
Design changes: May be referred to as “engineering change” or “enterprise change,” this process identifies and documents any design changes.
Design history file: This is the formal document prepared for each medical device that includes either all documents generated in product development process OR an index of documents and locations where they are stored.
As you can see, the DHF is one of many design controls, but it also serves as the repository of records of all other design controls. Think of it as the final stop for the information generated by all other design controls.
What is in a Device Medical Record (DMR)?
The third leg in the tripod supporting your compliance process is the Device Medical Record, or DMR. Think of the DMR as the “recipe” that resulted from the design controls to manufacture the device according to design specifications. It’s more than just a production traveler—it will include device specifications, compositions, drawings, formulations, software specifications, etc., as well as production process specifications, including appropriate equipment, methods, procedures and environments, such as clean rooms, humidity controls, quality assurance procedures, and any specialized equipment needed. The DMR will include everything from beginning to end of the production and shipping process, including any relevant installation instructions.
What are some pitfalls to avoid when compiling the DHF?
Many companies make costly and time-consuming errors when creating their DHFs, leading to longer-than-necessary audits and failed FDA inspections. Avoid these mistakes, and you’ll minimize lost time and unnecessary costs getting to market.
Jama Software hosted a webinar led by Quality Centric Consulting to review:
The top DHF errors that are costing you money
How to tackle 21 CFR 820.30 requirements
How Jama Connect™ streamlines the DHF process
Five of the most common pitfalls of the DHF include:
Not having a DHF: Usually small companies strapped for resources will put all their efforts into designing and getting a product to market, with the intention of doing the DHF later on. This could be due to a lack of understanding of what DHF outputs are required.
Chaotic and unorganized DHF: This is more typical in paper-based companies that keep all information in binders, but leave off dates or include unapproved documents, making it impossible to organize at the time of an audit or inspection. Disorganization — whether paper- or email-based — also makes it difficult to manage changes and revisions due to information silos and lack of sharing information.
Including unnecessary documents: Adding business documents, such as supplier quotes, financial information or unused concepts, can bring a flurry of unwanted questions from inspectors. This can also be a symptom of not having a DHF tool or template, as well as a linear process that involves lengthy review and approval times.
Unmaintained DHF: Often companies use either outdated or improper technology for the job. Paper-based systems, email, shared folders with read-write access and the like, are not always effective document controls. This can lead to lost copies of important records, making a DHF non-compliant.
Not traceable to outputs: Without a DHF, it’s impossible to create your DMR, and very difficult to establish your traceability matrix. If design control results are not well documented, easy to locate or current, your DMR will fail to be compliant.
Creating the DHF may feel like a heavy burden, but it is the key to a smooth compliance process. By treating the DHF as part of your design and development process from the beginning, these requirements become seamless and help ease the path to compliance.
Jama Connect can help traceability and requirements management and encourage a systems approach to medical device development, making the creation of a DHF more seamless and streamlined than ever before. To learn more, contact Jama Software.
Over the last decade, digital health solutions have become increasingly important, powering revolutionary developments in areas such as robotics, genomics, diagnostics, patient/provider interactions, and many more.
In our recent webinar with MDM Engineering Consultants and BeanStock Ventures, we talked about critical standards and best practices to follow. Attendees also heard critical insight from industry leaders in digital health development.
Watch the webinar or read the recording to learn more about:
Digital health solution classifications
The most common digital health development challenges
How Jama Connect can be leveraged in your development process to help ensure FDA compliance
Industry leading insight and knowledge
Below is an abbreviated transcript and a recording of our webinar, Ensuring FDA Compliance for Your Digital Health Solution.
Mercedes Massana: Digital health merges different digital technologies to make health and healthcare applicable to our living in society and make it more personalized for the patient. Many people see digital health as the future of healthcare. Some things that are digital health are like wearable devices. For example, your Apple Watch. It has some sensors that can do an EKG. That would be considered a wearable device. Things like telehealth and telemedicine, which we all became more familiar with because of COVID, are also part of digital health.
For digital health to use technologies such as different computing platforms connectivity, which is very important because you’re trying to get information to the healthcare provider, software plays a huge and significant role in digital health, and then different technology with sensors. So these technologies are intended to be used as a medical product or as a companion diagnostic to a medical product and help the healthcare provider make decisions. Software is playing an increasingly important role in healthcare in general. As medical devices have gotten more complex, software has played a bigger role.
Three Types of Software in Medical Devices
Mercedes Massana: There are three different types of software related to medical device. One is software in a medical device, embedded in a medical device. Another is software as a medical device, which we’ll call SaMD, and we’ll be discussing today. And the third is software used to produce or maintain a medical device. All these three types of software are regulated by the different regulatory agencies. Software as a medical device, is software that is intended to be used for one or more medical purposes that perform these purposes without being embedded in a medical device. So when we say embedded in a medical device, it’s software that’s physically part of a medical device. Like an infusion pump has embedded software in it that runs on the GUI on infusion pump.
This means that this software that’s a medical device in itself is not something that’s tangible. It’s not something that you can touch, like a medical device where you can touch. Well, this software lives on the ether and it’s not something you can touch. But it provides meaningful information that can help you mitigate disease, it can help you diagnose screening. For example, if you had software that took information from an MRI and helped define whether cancer was present, that would be software as a medical device. It takes information from another device and it helps a healthcare provider and make a diagnosis.
Determining the Intended Use of Software in a Medical Device
Mercedes Massana: So for software as a medical device, an important part is to determine what that intended use is. And in order to do that, guidance documents provide… You have to provide the answers to these three very direct questions. One is what is the significance of the information provided by SaMD to the healthcare decision? Is this information going to be used to help treat a disease, to help diagnose a disease, to help clinical management of the disease like diabetes, or to just inform clinical management? Obviously, it’s more critical if it’s going to treat or diagnose a disease. We also want to know the healthcare situation or condition, the scenario in which the software is going to be used and whether absence for incorrect information at that time can result in critical and putting somebody in a critical position, where they might be subjected to serious injury. Or in a serious position where they might be subjected to additional procedures and things that would not be necessary. Or in a non-serious position where it’s just information is not really that critical.
And then we also want to know what is the core functionality of the software. What are the what is it supposed to do? What are his critical features and functions? So based on these three questions, we would craft an intended use statement for the software as a medical device. The healthcare situation, there’s three different classifications, as we said. Critical is where accurate or timely diagnosis or treatment action is vital to avoid death or serious injury. Serious is where we need accurate and timely information in order to avoid an unnecessary intervention. And then non-serious is where the information is important, but it’s not critical in order to avoid that serious injury or unnecessary intervention.
Using Categories provided by International Medical Device Regulators Forum (IMDRF)
Mercedes Massana: Once we’ve determined what that healthcare situation or condition is, then we can determine the significance of the software. We use the categories provided by IMDRF, which is the International Medical Device Regulators Forum. This organization has actually generated some guidance for SaMD. And it’s used by regulators to try to have some common level of standards on how the software is going to be regulated. So in order to get to the classification, we determine the healthcare condition, what’s that healthcare scenario. And then we determine what that information provided is going to be used for. So if it’s going to be used to treat or diagnose disease, and it’s in a critical scenario, that will have the highest level of criticality, which is a four. There’s four levels of classification one being the least critical.
Again, it’s a level of one to four. Four has a very high impact and one in low impact. So, a device that would screen for cancer or something that software would be used to screen for cancer, would be very high impact. Something where maybe we’re sending a heart rate for somebody who’s going through cardiac rehabilitation, that might be a lower impact because it’s just using to inform the clinician on how things are going, but it’s not being used directly to impact on the treatment. There are additional ways to classify medical devices. The global harmonization task force has a classification of A, B, C, or D. So four stage classification, where A is the lowest level of risk, and D is the highest level of risk. These can be used because SaMD is in itself a medical device, we can use this level too.
Mercedes Massana: Additionally, IEC 62304 classifies software in three different classes, C, B or A, where C is if the software fails, it can result in serious injury or death. B it can result in non-serious injury. And A would result in no injury or harm. Now, why is it important to classify software? Because this will tell us how much effort we need to put into the documentation and testing and so forth of software. Obviously, the more critical it is, the more effort it will be required. FDA also classifies software. They have a level of concern for software. And there’s three levels also which align pretty well with the definition of IEC 62304. For FDA, they use major, moderate and minor.
Software in general has always been challenging for manufacturing. Software as a Medical Device (SaMD) poses some additional challenges to the developer of the software. Some of these are, for example, the use of off-the-shelf software is a lot more prevalent in SaMD than it is with embedded devices. Regulatory agencies want to see a lot of information when you’re using off-the-shelf software or suit, which is software of unknown providence, so there’s a lot of requirements that you need to meet. This is essentially software that you don’t own, you don’t have the source code for, but it’s still used in your medical device, so you’re still responsible to ensure that it’s safe within the parameters that you’re using it. Software as a Medical Device also, it will run on different platforms. It can run on a computer. It can run on an iPad, it can run on a smartphone as opposed to embedded software where you know the environment that it will always operate in. So it is a challenge to ensure that software behaves effectively in any of the platforms that it will run it.
Challenge: Interconnectivity with Other Systems
Mercedes Massana: Even though most medical devices today have some form of interconnection right to other systems, for SaMD, it’s almost given that it will have to be interconnected because the information will have to get to the healthcare provider, and that is typically through the internet. It’s going to be sent… There will be connectivity to other systems that will provide challenges like cybersecurity. We need to keep information safe. Those are challenges that we need to meet. Secondly, the method of distribution, the frequency of updates of SaMD is different than that of an embedded medical device. So for an embedded medical device, the device gets built, the software gets put in EPROMS that gets built into the device, the device gets packaged. It gets sent out to a pharmacy where you might buy it or it gets sent out to a hospital where it will be unpacked and tested.
Well, Software as a Medical Device is not a tangible thing, so it’s going to be distributed through the internet there is no checks and balances there. There’s no checksum to know that you’ve downloaded it correctly, so it proves some additional challenges there. And then lastly here, it’s much easier to duplicate software that’s on the internet that you can download. The manufacturer doesn’t always have control of how this software is duplicated, as opposed to software for an embedded medical device where in the manufacturing facility it gets thrown into EPROMS and then gets put into the medical device. So the manufacturer has control. For SaMD, the manufacturer does not have control, or does not always have control.
Mercedes Massana: For most applications, the apps we have on our smartphones, they get updated probably three, four, five, six times a year. So for a medical device software, you need to have a pretty rapid development time in order to keep up with this change of information. Then there’s also cybersecurity risks that we need to be aware of. Again, information security is a big issue. SaMD is something that regulatory agencies are very interested in. How do we get to meet these development challenges? Well, we need to have a very robust design development process to help us meet these challenges. We need a process that’s very methodical and systematic. We need a good quality management system. And then we need a software lifecycle process that takes us through the entire software lifecycle for the development of this application. It should take us from the point of where we’re starting to understand user needs, all the way to where we’re ready to retire the software. IEC 62304 is such a process where most people that are developing medical device software are utilizing this process.
Challenge: Managing Software Changes
Mercedes Massana: In addition, because the environment that the software is used and can change after release of the software, it is important to have a very strong post-market surveillance process to monitor customer issues in the field. You also need to monitor for cyber security threats, things like that. And you might need to make changes to your software based on what you’re observing in the field. So a post-market surveillance process is very important. Additionally, managing software changes, which is always important no matter what the software is used for, whether it’s a Software as a Medical Device, or embedded software in a medical device, is very important to manage changes. But for Software as a Medical Device, it’s even more important because what you change in the software could actually change the classification of the software. It could be more critical or less critical and it can also change the intended use. If you change the intended use, that means that you’re going to have to submit to regulatory agencies, so you want before you get approval to market that software.
There are a lot of software applications. Now, Software as a Medical Devices where they’re getting so much data and information that they’re using artificial intelligence, or machine learning to make the software smarter and better. Using this could potentially change the classification of the software. SaMD is still fairly new to regulatory agencies so I don’t think they’ve figured out exactly how they’re going to regulate things like artificial intelligence and machine language. But managing changes and understanding the impact of those changes is very important for our Software as a Medical Device. There aren’t a lot of guidance documents out there for Software as a Medical Device, but luckily there are a few standards that can guide us in our development efforts.
Behind every successful product and product launch lies a complicated process that can involve multiple oversight agencies, cross-functional teams, and stakeholders. Within an organization’s governance, risk management, and compliance (GRC) framework, product teams must pursue market innovation while remaining in compliance. Staying in compliance with the vast array of rules, procedures, and contract clauses that govern product development can be a full-time job, and lack of management can lead to delays and failures that result in lost revenue, damaged reputation, or even legal action.
With so much on the line, every development project should have a process within the GRC framework to monitor compliance risk and compliance activities along the way. Unfortunately, compliance management can be difficult to integrate into the full design process, and compliance delays can hold up a product launch. Product teams often need a better way to integrate compliance management into the development process. Those teams that include compliance management in their processes can reduce the risk of failure—and potentially improve their speed to market.
What is compliance management, and how does it help product teams ship on time?
Compliance can refer to a variety of different laws, guidelines, standards, processes, procedures, or other controlling documents, including contracts. Compliance management refers to the intentional process by which designated team members or compliance officers monitor and control design, development, launch, and fulfillment to ensure that all legal, contractual, and procedural requirements are met.
Ideally, compliance management involves both people and a system that is fully integrated across functions of the organization. The more integrated the compliance management in the development lifecycle, the more likely it is to detect potential compliance issues before they cause long-term harm to companies or consumers.
A fully integrated compliance management system can help your product team ship on time or early by keeping all compliance factors top of mind during the design, development, and fulfillment process. When teams are ensuring compliance throughout the entire development lifecycle, unforeseen delays and consumer issues are less likely to derail product success.
To fully integrate compliance management, teams need a development process that includes touchpoints for assessing compliance. The process should include activities such as:
Standard operating procedures
Safety and security procedures
Reporting and documentation
Internal and external audits
Integrating all of these activities will help ensure that teams are fully prepared to establish compliance with applicable standards, regulations, and laws before launch, reducing the possibility of costly delays or failures.
Compliance control is a set of guidelines and policies designed to provide a framework for compliance to product teams and stakeholders. These guidelines and policies apply to everyone involved in compliance management—from board of directors to compliance officers or compliance managers to team members.
Common internal compliance controls can include the following:
Published standards, policies, and operating procedures
Training and documented completion of training
Internal audits
Contracts
External compliance controls are those that originate anywhere outside the company:
Laws and regulations
Industry standards
External audits
External risk assessments
What is a compliance management system?
A compliance management system is a program that integrates written documents, processes, functions, controls, tools, and anything else that helps organizations comply with regulations and reduce risks to consumers that arise due to violation of applicable law. While a comprehensive compliance management system will include appropriate tools such as software, it will also clearly define the roles of various stakeholders, including:
Team members
Compliance officers or compliance managers
Board of directors
Internal auditors
The compliance management system will also define processes for:
Assessing and responding to consumer complaints
Addressing results of a compliance audit
Providing relevant compliance training as appropriate
Keeping informed of regulatory change
Taking corrective action when products are found in violation
Why is maintaining compliance so important for product teams working in regulated industries?
Former US Deputy Attorney General Paul McNulty has stated, “If you think compliance is expensive, try non-compliance.” In any industry, non-compliance can lead to fines, product failures, and lawsuits—not to mention the cost of lost reputation, customers, and business.
For teams working in regulated industries, maintaining compliance is especially important as there are often unique safety and regulatory issues at hand. In industries such as automotive, aerospace, and medical devices, one product that fails to meet regulatory or legal standards could potentially lead to the kind of product failure that results in loss of property or human life.
What are best practices for compliance management?
While the specific practices for compliance management will vary according to industry, there are some best practices for compliance management that any product team can use to ensure a successful product launch.
Identify compliance officers: If your company doesn’t have a dedicated compliance officer, identify someone on your team who can at least serve as an ad hoc compliance officer. Identifying the right person (or people) early in the development process will give a central point of contact so that no one is wondering who is in charge of compliance.
Learn relevant requirements: All members of the team should at least have a familiarity with the regulatory and legal environment around product development. In addition, everyone should subscribe to government and industry mailing lists or websites to stay up-to-date on changes to regulations. One study found that regulatory monitoring saved companies an average of $1.03 million.
Create a central repository of requirements: Rather than simply documenting requirements that are relevant only to one team or project, companies should create a central source of information for all compliance requirements, including internal procedures and standards. With one central source for all teams to access and contribute to, teams are less likely to miss requirements in the development process.
Establish traceability between requirements and standards, regulations, and other relevant compliance documents: By documenting traceability between the compliance requirements and project artifacts, product teams create a robust analysis tool that can provide invaluable detail and information for audits and reviews.
Implement tools that support compliance management: While there’s no substitute for having human input and control over compliance management, there’s no question that having the right tools can make compliance a more manageable task. The right tool will support product teams by providing a central source for traceability, requirements management, analysis, documentation, and all related activities.
What makes a compliance program effective?
A comprehensive and effective compliance program will:
Reduce costs: When teams have a program in place for comprehensive compliance management, it’s almost inevitable that the team or company will save money overall.
Improve risk management: Evaluating and assessing risks throughout the lifecycle will improve risk management across the development lifecycle. Good compliance management will help teams identify and assess risks early in the development process.
Keep product development well within organizational GRC framework: An effective compliance program will keep development in compliance with internal controls and the overall organizational GRC framework.
Improve speed to market: When compliance is fully integrated into product design and development, the risk of delays drops, and chances of shipping on time or early increase.
What tools and software can help product teams maintain compliance during the application lifecycle?
A compliance management solution is an important piece of any compliance management system. Within the overall framework of a compliance management system, a compliance management solution will support workflows, self-assessments, surveys, and issue remediation. In addition, intuitive dashboards and charts can provide real-time insights into compliance processes.
How can Jama help teams working in highly regulated industries, like medical and aerospace, maintain compliance during the application lifecycle?
Jama Connect gives product teams the tools they need to integrate compliance management into the design lifecycle. By tracking requirements and giving teams the traceability they need, Jama Connect simplifies compliance management by integrating it fully into the application lifecycle. To learn more about Jama Connect, contact us for a demo.
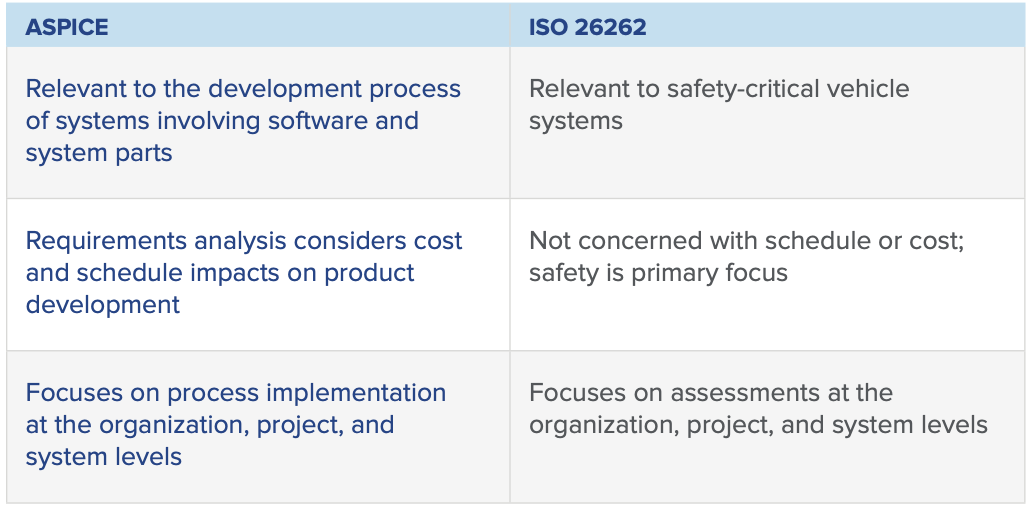
If you haven’t already, check out Part I of our ASPICE 101 blog series to learn about what the standard is and why it’s important to automotive development. In this post, we take a look at ISO 26262 vs. ASPICE and examine the similarities and differences between these two important automotive standards.
ISO 26262 vs. ASPICE for Automotive Compliance
Of course, automotive companies already use ISO 26262, and introducing yet another automotive compliance piece into a very full process may feel overwhelming. It’s understandable why companies would be asking if they need to adhere to both ASPICE and ISO 26262 when they are already focused on ISO 26262 compliance.
The answer, in short, is that while there is no regulatory requirement to use ASPICE, using the model can greatly benefit companies that want to stay competitive in the automotive industry. According to the Project Management Institute, 47% of project failures can be traced back to poor requirements; any guidance or set of standards that can help mitigate that risk is worth the implementation effort.
While ASPICE and ISO 26262 are complementary and do overlap in places, they ultimately serve different purposes. ISO 26262 covers functional safety standards for vehicles. It incorporates safety analysis methods that account for random and systematic errors in electrical and electronic systems and is broadly adopted worldwide. ASPICE is the current standard for software best practices in the automotive industry. It covers how to conduct software and systems design whether or not safety is a concern.
The best approach for automotive development teams is to consider both ASPICE and ISO 26262 guidelines. Below we will give a brief overview of both standards and discuss the similarities and differences.
ISO 26262 Explained
ISO 26262, titled “Road vehicles – Functional safety,” is an international standard for the functional safety of electrical and electronic (E/E) systems within road vehicles. Originating from the more generic IEC 61508 standard for electrical/electronic/programmable electronic safety-related systems, ISO 26262 addresses the specific needs and challenges of automotive E/E systems safety lifecycle management. This standard aims to ensure that E/E systems in vehicles are designed and developed to meet stringent safety requirements, reducing the risk of failures that could lead to accidents and harm.
The ISO 26262 standard is structured into several parts, covering aspects such as vocabulary, management of functional safety, concept phase, product development at the system, hardware, and software levels, production, operation, service, and decommissioning. It also includes guidance on automotive safety integrity levels (ASILs), which are used to classify and manage the safety requirements necessary to mitigate risks to an acceptable level.
Key aspects of ISO 26262 include:
Risk Analysis and Management: It emphasizes the identification, evaluation, and mitigation of risks associated with E/E system failures throughout the vehicle’s lifecycle.
Systematic and Random Hardware Failures: The standard addresses both systematic failures (due to errors in specification, design, manufacture, etc.) and random hardware failures, proposing methods to manage and mitigate their effects.
Functional Safety Assessment: It requires a structured functional safety assessment to be conducted at various stages of the product development process, ensuring that all safety goals have been met.
Automotive Safety Integrity Levels (ASILs): ISO 26262 introduces ASILs, which are assigned based on the severity, exposure, and controllability of potential hazards. ASILs range from A (lowest) to D (highest), dictating the rigor of safety measures needed.
Safety Lifecycle: The standard outlines a safety lifecycle for the development of automotive E/E systems, including specific processes and tasks that must be followed to achieve functional safety.
Documentation and Evidence: Comprehensive documentation and evidence of compliance with the standard’s requirements are critical for the certification process, supporting the safety case of the E/E system.
ISO 26262 is applicable to all types of passenger cars, motorcycles, trucks, buses, and trailers, with its principles also being adapted for use in other automotive applications. The standard is continually evolving to address the advancements in automotive technologies, such as autonomous vehicles and electric mobility, ensuring it remains relevant and effective in managing functional safety in the dynamic automotive industry.
ASPICE Explained
Automotive SPICE (Software Process Improvement and Capability dEtermination) is a framework used within the automotive industry to assess and improve the maturity of software development processes. It is based on the ISO/IEC 15504 standard, often referred to as SPICE, and tailored specifically for automotive software development and related system integration processes. The framework is designed to help organizations develop high-quality automotive software more efficiently, ensuring that it meets both customer expectations and regulatory requirements.
ASPICE provides a structured approach to evaluating the capability levels of an organization’s processes in a consistent manner. It defines a set of process assessment models and practices that organizations can use to measure their processes against industry best practices. The framework focuses on key process areas such as software engineering, project management, quality assurance, and supplier management.
Key features of ASPICE include:
Process Reference Model (PRM): This model defines the processes considered essential for the development and management of automotive software. Each process is described in terms of its purpose, outcomes, and outputs.
Process Assessment Model (PAM): The PAM provides criteria for assessing the maturity levels of the processes defined in the PRM. It outlines capability levels (ranging from 0 to 5) and process attributes that are used to evaluate the performance and capability of processes.
Capability Levels: These levels describe the maturity and capability of processes within an organization. They range from Level 0 (Incomplete) to Level 5 (Optimizing), with higher levels indicating more mature and capable processes.
Assessment and Improvement: ASPICE not only enables the assessment of current process capabilities but also provides a framework for continuous process improvement. Organizations can identify gaps in their processes and implement targeted improvements to enhance their software development capabilities.
ASPICE assessments are typically conducted by certified assessors who evaluate an organization’s processes against the framework’s criteria. The outcome of an assessment can help organizations identify areas for improvement, increase the efficiency of their software development processes, and enhance the quality of their automotive software products.
By implementing ASPICE, organizations in the automotive industry can achieve several benefits, including improved process transparency, higher software quality, reduced development risks, and better alignment with industry best practices. As automotive systems become increasingly software-driven, adhering to frameworks like ASPICE is becoming more critical for manufacturers and suppliers aiming to meet the high safety, reliability, and performance standards expected in the industry.
ISO 26262 vs. ASPICE: Similarities and Differences
There are several key distinctions between ASPICE and ISO 26262:
Stay tuned for our next post in the ASPICE 101 blog series where we discuss goals, requirements, and levels of ASPICE compliance.
Editors note: This post was written partially assisted by artificial intelligence. It was reviewed for accuracy by McKenzie Jonsson and Deco Wilkerson.
2020 has been a year that’s been described as “unprecedented” and “unparalleled” – as well as other descriptors probably best left out of our blog. As we close out this year, it’s hard to say what awaits us in the new one. One thing that we can be sure of is that innovation in medicine, science, and technology shows no sign of slowing down.
As we enter a new year of technological advancements, Jama Software asked select thought leaders – both internal and external – across various industries for the trends and events they foresee unfolding over the next year and beyond.
In Part I of our four-part series, we ask Steven Meadows, Medical Device Solutions Lead at Jama Software, and Founder & CEO, Shawnnah Monterrey at Beanstock Ventures to weigh in on trends they see in the medical device industry for 2021.
Note: Now that our 2021 Predictions Series is complete, you can also go back and read, Part II,Part III, and Part IV.
What are the biggest trends you’re seeing in the medical device industry right now? How will they impact products, systems, and software development?
Steven Meadows:
From a product perspective, there are a number of areas in the medical space that are set to grow in 2021 and beyond, including robotics nanotechnology, wearable health tech, and extended reality devices.
With more medical devices incorporating software components and connecting to systems, like hospital networks, enhanced cybersecurity is becoming increasingly important. Medical device companies will continue to invest heavily in this area through 2021 and beyond.
Shawnnah Monterrey:
There are three primary trends that I am seeing: A shift in initial product launches from Europe to the United States; An increase in Software as Medical Device products; and an increase in IVDsfrom lab-developed tests.
We are seeing a shift in initial product launches from Europe to the United States, due to the upcomingEU MDR regulations.Typically, US–based companies would launch medical products in Europe first, followed by a US launch shortly after due to the rigor and lag time in receiving FDA approval. CE Mark was typically more straightforward, required less clinical evidence, and was a faster process, especially when companies could self-certify.EU MDR is currently being perceived to be more onerous than the FDA regulatory clearance process and is therefore creating a trend of product launches occurring first in the United States and then in Europe. The cascading impact would therefore be the need for more services providing expertise in regulationsto support EU MDR and a need for a more harmonized regulatory strategy.
The second trend we are seeing is an increase in Software as a Medical Device (SaMD). There have been numerous healthcare, fitness, and consumer products launched into industry recently that were not previously considered a medical device. As a result of the SaMD FDA guidancecombined with the EU changes from CE, MDDto EU MDR, many products that were not originally deemed clinical products or medical devices are now falling into that category. Products that were previously not classified as medical deviceswere not necessarily held to the same standards thatare required to produce a medical-grade product such as quality management, design control, risk management, clinical studies, and post-market surveillance. This shift will cause companies to delay product launches in order to satisfy the regulatory requirements, resulting in an increased need for training, systems, software, and tools to make it easier for companies to make that transition. Since these companies generally lackquality and regulatory expertise, additional support will be requiredto deliver medical-grade software products.
The last trend we are seeing is an increase in IVDsfrom lab-developed tests (LDTs), research use only (RUO) and emergency use authorization (EUA).There are numerous laboratory-basedtests and associated software that havebeen developed to support very complex assaysthat are being used clinically which has recently been deemed outside of the scope of LDT guidance.Products that were covered under CLIA are now required to be cleared with the FDA and EUas an IVD.From a CLIA perspective, the focus is on validation. Validation is centered on the intended use of the product and workflow. But with medical devices such as IVDs, there needs to be a shift to bringing it down to verification levels –namely design verification. Design verification is quite different than validation, design verification islooking at the design aspects of the product, not from a workflow perspective, but from a design feature perspective.Design verification occurs at the subsystem and system level, where the subsystem is the breakdown of the system from the architecture.The scrutiny will be from a hazard perspective for the entire system and a failure mode analysis perspective at the subsystem level. Finally, ensuring that there is a full product definition and full verification associated with theproduct. The impact is that it is going to require taking a different look at the software that has been provided and looking at it differently from a process software perspective to an IVD perspective this might require looking at the architecture and redesigning the system to break apart the items that were allocated to CLIA and the lab system processes versus the actual product that performs the clinical interpretation and the diagnosis.
An overarching impact will be evolving medical device productdevelopment from a methodology perspective. Manycompanies are lean and agile and are performing continuous integration and development of their products. It will be important to set up processes and procedures to still allow for that while continuing to meet the regulations. From a systems perspective, it would be beneficial to take advantage of available tools.There isa myriad of tools in place that can assist indevelopingquality management systems, manage design control, support requirement and risk management, testing, test automation, and deployment. There is anopportunity for companies to start investing in tools to help withcompliance and save time and money whilestill maintainingtheir efficiencies.
In terms of product and systems development, what do you think will remain the same over the next decade? What will change?
Steven Meadows:
Software is becoming an increasingly used component in medical devices and we expect this trend to continue. More of our customers are adopting an Agile methodology when developing their software, a shift that has been happening for some time, and will continue to over the next decade.
Shawnnah Monterrey:
Over the next decade, I see the use of Artificial Intelligence (AI) supportingthe creationof sophisticated data-drivenalgorithms such as autonomous diagnosis. AI is gainingrenewed traction, even though it has been used in medical products since the 2000s. I believe there will continue to be a tremendous amount of investmentin the development and understanding AI in the healthcare space and how it could be used to improve clinical outcomes. AI will continue to progress over time as the industry develops an understanding of the utility and how it can contribute to guiding clinical decisions as an autonomous tool. Thereis FDAdraft guidance around AI, however, I think the biggest potential constraint will be how to continuously improve upon the algorithms as new data comes in, without requiring a new regulatory submission.
The other trend I see progressing over the next 10 years is one we are already seeing,cybersecurity. This trend is a bi-product of the increasing numbers of clinical cloud applicationsthat integrate with or use medical devices. As the need for sharing clinical data, advanced computing, remote diagnostics,and accessibility increases, so will the migration of healthcare applications to the cloud.This increased need will be the catalyst that pushes up the demand for securing patient data not only in terms of patient privacy but also in terms of patient safety.We have seen several instances now where, established medical device manufacturers, medical device cloud software, could be hackedby an unauthorized person and potentially cause adverse side effects to the patient. Such hazards could trigger a class I recall, the most serious type of recall. Not only do these hazards put patients at risk, but they also impact a company’s reputation and financial viability. Occurrences like this will justifiably result in a growing emphasis on creating software systems, processes, and tools tosupport the industry inmaturing standards and guidance around cybersecurity.
In terms of what will remain the same in the medical device industry throughout 2021, I would say that the regulationswill remain unchanged. It is unlikely we will see a change in this coming year. To the contrary, we will likely see an increase in the regulation. Ipreviously discussed the potential for harmonization. However, I do notbelieve that will happen until there is a push for it or until the FDA acts on the overlap between many of the guidance documents. Additionally, there will need to be harmonization on the development methodology across different devices so there is not a need to have different guidance documents. In short, I do not see the regulations changing over the coming year.
In discussing 2021, we should also take a moment to discuss Covid-19. It’s not my belief that things will change from a Covid perspective. The trend that I see continuing involves businessesincreasingly going digital, putting pressure on products to focus on cybersecurity. Specialties, such as telemedicine will likely increase, requiring true patient authentication and there will be progress, but a delay in medical device development in areas of selected surgeries.
Regarding development teams, I think Agile is here to stay. I thinkthat development teams will continue to use the agile approach and methodology. The organizational struggle of internal alignment of the quality management system will persist until there is adisruption in the regulatory environment itself. Relative to the need for systems as software tools become more useful and more integrated. The main area I see increasing are the new entrants into the market.There has been a large amount of new investment and I believe that will continue.I think we can assume that there will still be quite a bit of investment opportunities for new players.
What sorts of process adjustments do you think development teams will need to make to accommodate these changes? Do you think they’ll need to make technology investments, process adjustments, or both?
Steven Meadows:
As we continue to deal with the new normal, I expect medical device teams to continue to invest in collaborative product development software, enabling their teams to continue bringing new devices to market in a safe and competitive way. Development teams, using legacy product development platforms and Word/Excel need to switch to a modern alternative, in order to be as successful as possible in 2021.
Shawnnah Monterrey:
My belief is that development teams are going to need both technology investments and process adjustments to accommodate the changes that are coming. There will be a heavy reliance on technology as the software realm moves more towards focusing on core IP and leveraging the existing software components available. For example, there could be an organizationsuccessfully providing cybersecurity packages where other organizations may not have that expertise, so they would be leveraging assets from another organization.In short, software re-use will be important.
My sense is that the industry will shift more towards investing in technology such as AI and mechanisms to improve data sharing.There is an opportunity to really understand the clinical value that the data brings.Currently, the healthcare environment iscomprised of disparate systems that will eventually need to come together to be able to fully leverage the data and use the data in unique yet meaningful ways.
We are currently seeing a big shift where the medical device companies and their quality management systems do not satisfy the way in which engineers perform medical device development. For example, software engineers are very agile and tool oriented by nature, while many established companies still utilizea paper-based quality management system or document control system. We’d like to see software teams and the Agile methodologiesdrive changes to the quality system so that the quality system can be nimbler and more adaptiveallowing them torelease compliant products faster.
We have observed a hindrance in the companies and auditors themselves to adopt a more progressive and nimble approach that is not necessarily driven by the regulations. Our interpretation is that the intent of the current regulations is to be nimble andstill provide safe and effective products.My belief is that if we do not make a change to the current way in which we develop medical devices in thecorporate environment, innovation from these larger organizations will be stifled. The innovation gap will ultimately be filled by startups entering the marketplace and experiencingimmediate success because the old way of doing things is no longer sustainable or effective. I foresee a trend in an increase in the volume of new, emerging, small players. These players are going to be nimble and will look at the regulations from a different perspective.
How do you foresee regulations shifting in the medical device industry over the next decade?
Steven Meadows:
Key medical device standards and regulations are constantly changing. Notably, EU MDR is undergoing a number of changes and will need to be fully implemented by May 2025. Key standards like ISO 14971 go through multiple iterations and Jama can help our customers stay current with these changes.
Shawnnah Monterrey:
I will say something that ‘should’ happen, not that ‘will’ happen,relative to regulatory changes in the medical device industry over the next decade.Ideally, there would be harmonization across the guidance documents. There are too many guidance documents and many of them are repeating the same requirements but in different ways. With a harmonization effort, it would remove the perceived burden of applying multipleguidance and standard documents. With the current format, there is a need to understand each and every guidance and standard document andto be able to apply them all correctly. With more alignment and more harmonization, it would facilitate adoption and push forward innovation.
The impact of harmonization would certainly be in facilitating development. Withone cohesive guidance document and less confusion in applying multiple regulations and guidance documents across development, it would result in more development. If there was more alignment across the agencies to create cohesive processes, it would be easier to get consistency in the application of it. Additionally, the FDA’s time to approve products would be reduced because the FDA would be seeing similar submissions and be able to focus on the critical aspects of the product.
Anotherregulatory change that I expect to grow over the next decade is with the FDA’s adoption of third-party review organizations. The FDA has begun utilizing third-party vendors to streamline and offload their burdenof reviewing Class I & II devices. By working with approved partners to facilitate the review, the FDA can focus on higher risk devices. This allows a company’s submissionto get reviewed more quickly. Over the next decade, the FDA will likely become more sophisticated in this partnership with third-party organizations. The impact would be a faster time to market, possibly launching products months more quickly.
What do you think will be some of the differentiators between a company surviving to see 2030, and those that do not?
Steven Meadows:
Medical device companies that embrace a proactive approach to quality will ultimately find fewer issues with their products, improve customer satisfaction, and stay competitive for the foreseeable future. Companies that invest in best-of-breed medical device development solutions like Jama Connect will have the upper hand in reducing risk and complying with various standards and regulations.
New cutting-edge technologies in the medical field, like robotics nanotechnology, and wearable health tech devices, bring complexities for medical device companies and risk for patients and consumers. Jama Software will continue to serve as the leading solution and partner to help innovative companies bring medical products to market, in a collaborative, safe, and efficient way.
Will those trends still be prevalent 5 years from now? 10 years?
Shawnnah Monterrey:
I believe there will be disruption in the regulatory space in the next five to ten years, so I do not see the trends staying the same.
It will be important to have industry guidance by having industry experts partner with the FDA to help make those improvements. Based on the FDA approach currently, I believe they will welcome industry guidance.In relation to the previous discussion of the FDA 510(k) Third-Party Review Program, if we want to put it into a broader bucket, this is really the FDAutilizingexternal resources to support the industry.However, the collaboration is not simply due to a lack of resources, it is also because there is a very challenging, difficult infrastructure to navigate. I believe that there is a push to simplify the system and there will be a reliance on industry partners to do so. The industry partners will include those that are doing software product development, providing quality regulatory services, and providing tools to help facilitate medical device development.
What innovations in regulation in the medical device industry do you hope to see in 2021? 5 years from now? 10 years?
Shawnnah Monterrey:
The innovations in regulation in the medical device industry that I hope to see is the increasingopportunities for companies to collaborate with the FDA. I also hope to see more companies, larger organizations, or startups, take a more lean, nimble approach and be more receptive to the Agile methodology that software development teams are currently using. Current processes and procedures were generally derived from good engineering practices;however,perspectives of quality and regulatory experts and independent auditors remain the same. I would like to see them be more adaptive towards compliance,as compared tothetypical approach of checking the box, and instead, establish a partnership with their medical device development team to help facilitate compliance without sacrificing the quality of the product.
More specifically, I would like to see more efficient uses of tools and technology to support medical device development. There are many tools out there, butthey are very disparate.I believe that with more cohesion between those tools it would better facilitate medical device development to comply with the regulations.
In five years, my hope would be that there would be a first pass at simplifying the guidance documents.
In 10 years, I hope to see a streamlining of the processes. I hope for standardization of the submissions.I would hope to see a simplification of the current guidance documents across the board in the form of a standard method for supporting 510(k) and PMA submissions.
Stay tuned in the coming weeks for additional 2021 predictions! In the meantime, to see more information specific to the medical device development industry, we’ve compiled a handy list of valuable resources for you!
The new regulation, which emphasizes patient safety, traceability, and transparency, will impact medical device developers, manufacturers, and suppliers of all sizes across Europe, and will require fundamental changes throughout the entire device lifecycle.
In order to better understand the regulation and its impact on our medical device customers, we sat down with Satyajit Ketkar, Principal System Architect & Engineer at Velentium, to learn more about what MDR is, what medical device developers can expect, and what they should be doing to prepare prior to the May 26, 2020 implementation date.
Jama Software: What is EU MDR and what can medical device developers in Europe expect?
SatyajitKetkar:
MDR stands for Medical Device Regulation. It combines and replaces the existing directives. Once the MDR goes into effect (on May 26, 2020), the existing directives will be obsoleted and invalid. All medical device manufacturers will be required to comply to the MDR. Right now, there are three different directives: The Medical Device Directive (MDD), the Active Implantable Device Directive (AIMDD), and the In Vitro Diagnostic Devices Directive (IVDD) — The EU governing body took two of those —the MDD and the AIMDD — combined them together and brought it to the next level to make it a regulation, which means that it’s extremely explicit and binding. There’s also the IVDDR, like the MDR and will replace the IVDD. The MDR is also harmonized with a lot of the latest standards. EU MDR is supposed to provide the medical device manufacturers a much more comprehensive regulatory guidance on how to manufacture a medical device. But it goes way beyond that, it goes from phase-zero feasibility, all the way through end–of–life (EOL) of the device. There’s a lot of emphasis on clinical, post-market surveillance, how to handle complaints, auditing, review cycle – whether it’s three-year or five-year. There’s a lot more content in there. It’s a pretty large document. It’s supposed to be in the same line as the data protection regulation (GDPR).
Jama Software: Why are they implementing new regulations around medical device development?
Satyajit Ketkar:
They’re basically pushing these regulations up to a level where the ambiguity and the ‘legalese’ is taken out. In the past, a directive was set out through the EU legislative body from Brussels, and then each competent authority for each country would interpret it in their own way. Each of the notified bodies, like BSI, TÜV, or DEKRA, had their own interpretation of the directive. So, if you’re a legal manufacturer, you could go to each one of these bodies and get a different implementation of the directive. What this new EU MDR regulation is supposed to do is to clear that up. Everybody now operates in the same manner, in the same language. And because of that, the regulation is large.Whereas the MDD about 60 pages and has 23 articles and 12 annexes, the MDR is about 175 pages and consists of 123 articles and 17 annexes.
Jama Software:
What are the top things that you suggest medical device companies do to prepare for the new regulation?
Satyajit Ketkar:
Overall, one of the first things I would recommend any medical device manufacturer do is to read the articles that talk about device classification and understand where they fit in.
If you’re an existing manufacturer of a product that’s already out in the market that falls under either the MDD or AIMDD, you’ll need to understand where you fall on the new spectrum. You must understand if your medical device still falls under the same classification, or if it has changed.
Most likely, they’ve gone up in classification. If they were Class 1R or Class 1M, now they’re Class 2A. If they were Class 2A, they’re now Class 2B. If they were Class 2B, they’re now Class 3. And of course, the current or future AIMDsremain as Class 3.
So, what does that mean? That means that the rigor of their development processes and the evidence that they mustmaintain, the DHF (Design History File) records and what they mustprovide has just gone up by quite a bit. When these directives were originally written back in the ‘90s, a lot of the technologies we’re working with today didn’t exist. So, to capture a lot of that, they have up classified (changes to the rules for a device classification, see above).This is especially true for electrical hardware, firmware, software, cybersecurity, and the mobile applications.The rules and expectations for these areas have gotten more explicit and stricter.
It seems like a big part of MDR is that there’s going to be more traceability needed, unique identifiers, and more labeling for each device. Do you see that impacting the way organizations develop medical devices? And what’s the best way to capture that information, or anything else related to post-market surveillance?
Satyajit Ketkar:
So, for the UDI implant card, etc., that’s new, and wasn’t there before. So, everyone’s starting from scratch.
Some people were kind of doing UDI (Unique Identification Number) labeling in the Instructions for Use (IFU) or maybe the eIFU, but it wasn’t to this level of rigor. You must now maintain vigilant records for all of this, for the entire life of the product including part of the post-market. There’s supposed to be a whole database that they’re putting together to keep track of all the vigilant reporting and the post-market reporting, that’s called EUDAMED. There are now updates to the Med Dev guidance(s) on clinical and post-market that are going to be referred to within the regulation. So, I would recommend that manufacturers continue to follow and comply to that. Beyond that, it’s pretty much the same thing.
What’s changed significantly is the clinical follow-up. There’s a whole new guidance on clinical safety and performance that they must maintain, which is part of this UDI tracking and vigilant tracking. Also, there is a periodic clinical evaluationthat mustbe conducted. There’s aperiodic safety and performance report as well that needs to be updated thatmust be included in the vigilance and post-market surveillance. These, and the UDI, are all new with MDR. Before, you would do this once in your three or five-year review cycle, and then that was it. Now, you must basically maintain these all the time as part of your post-market surveillance.
This doesn’t introduce a development burden, but it is a lifecycle burden. You must maintain these throughout the life of the product, which could be 15, 20 years, especially if it’s an active implantable, a pacemaker, or any sort of a therapy device.
One of the other things that I would recommend on the regulatory side or the system side is to review Annex 1, which contains the safety and performance requirements called GSPR (General Safety and Performance Requirements). The MDD has a set of 13 EssentialRequirements (ERs), and the AIMDD has16. The MDR has 23 GSPRs. This might seem less overall but each one of these requirements has 10 to 30 sub-sections that you must adhere to. So, in reality, the requirements have more than doubled.
Typically, what happens is that when somebody in a regulatory group or clinical group puts together their submission package, they’ll review all these essential performance and safety requirements to make sure that they’ve got all the documentation or all the deliverables. They’ve gotten very explicit as to what you need to provide. One example, specifically Section 17.2, which talks about electronic programmable systems and software. They’ve gotten specific about cybersecurity. They’re catching up with the FDA on cybersecurity and software lifecycle. That’s just one example, but there’s a whole bunch of other ones in there. These are new. So, that is something that they need to be aware of as they do their preliminary risk assessment and overall risk managementto comply with ISO 14971.These will be additional inputs for decomposing their system requirements. They need to be aware of the new and existing requirements.
Learn how Jama Software can help medical device developers better manage risk with ISO 14971 by downloading our white paper.
Jama Software:
Do you think MDR will have an impact on the way people create and approve requirements? Do you have any thoughts on how the requirements managementprocess might change?
Satyajit Ketkar:
I’m moderately confident that when it comes down to a very specific phase(s) of the development lifecycle, where you’re decomposing requirements and creating your traceability, from design, implementation, through to verification, that it should stay the same. It’s just the details of what you’d have to maintain that are going to change. The process should stay the same. Because remember, there are two parts of what you do: You create a DHF to maintain for yourself as a manufacturer, and then there’s a subset of that that you send to a notified body. The burden of what you must maintain for your own records goes up quite a bit. The process stays the same. It’s just what you must keep track of now that has gone up. The traceability is going to go up a little bit, but the process of decomposing requirements to subsystem, technical, and doing the architecture design, etc., all the current and standard processes should stay the same.
Jama Software:
Do you see this impacting the way global companies communicate or collaborate as part of MDR?
Satyajit Ketkar:
Yes: it’s going to require quite a bit more communication. You must stay aligned a lot more closely because there’s a lot more burden. There’s a lot more meat on the bone you must provide. So, if you have a distributed team across multiple regions, you need a central repository to maintain all these documents.
Jama Software:
You mentioned the DHF and that burden of proof is going to really be on the company.Does it impact the way folks adhere to ISO 13485 or Quality Management Standards at all?
Satyajit Ketkar:
No, it should not. Since it is a brand-new regulation, one of the things they’ve done with the MDR is adopt all the latest harmonized standards. That includes the latest QMS standard, latest risk management, etc. So, if you’re up to date on those, then you should be fine. You should not need to change your QMS to adhere to the MDR as long as you are maintaining the state-of–the–art.
Jama Software:
You mentioned doing earlier risk management followingISO 14971. Are there any impactsto the risk management process?
Satyajit Ketkar:
No. It’s kind of the other way around. MDR has taken account for the updates to all these other standards, especially the updates to risk and QMS. So, if you follow the MDR and you’re up to date on your revision of these other standards, you should be fine.
Jama Software: Is there anything else that you think we missed, or you think should be top-of-mind related to MDR?
Satyajit Ketkar:
I want to be very specific about what I just said earlier in following the harmonized standards and maintaining the state-of-the-art. If you’re not up to date on the latest standards, for example, if you’re not following ISO 14971:2012, or if you’re not following QMS:2016, if you are behind and you want to jump up to the MDR, you’re going to have a pretty heavy lift because you must comply to the latest revisions as well. It’s all included in the harmonized standards list now as this list has been updated. So, if you’re already doing it (not just complying to the current harmonized standard but aware of the state-of-art), then you’re fine. If you’re not, there’s going to be some uphill climbing that may be required.
What’s typical in my experience is that people try to keep up with the standards more often than the regulations. But they’ll see a big jump in the regulation, and they don’t want to jump straight into the regulation and find out that they’re still back in 2006, they haven’t revved up to the latest one (typically the state-of-the-art), and they’re stuck. So, just want to make sure that manufacturers are aware of that.
Jama Software: How do you think a dedicated requirements management platform like Jama Software fits into all of this?
Satyajit Ketkar:
If you’re already using Jama Software, you won’t have to change the process too much. The only thing that this new regulation is going to change is that people are going to want to use Jama Software more because it’s going to become necessary. There’s just too much from a QMS perspective and a development process perspective to try to do manually.
Because of this new regulation, it’s going to slow down their development lifecycle at first, because they are now learning about this new regulation. On top of that, if you’re doing things manually, if you’re using Excel spreadsheets or Word documents, whatever it may be… You might have a very simple project, but now you must add that manual process burden on top of this new regulation. So, it would reduce that process burden greatly if you had a solution like Jama Connect that would handle a lot of this for you. If you could just enter in all your requirements into a lifecycle management tool that then allows you to create trace tables, allows you to create upward, downward decomposition, and presents that in a very clean way where you can export it to a DHF artifact. That saves time and effort.
That’s the whole point, is to save time, because you already are in a process and a regulation change.Tools like Jama Connect will accelerate that because I do foresee a major slowdown in the lifecycle of medical devices with this new regulation… especially for startups. They’re going to have a tough time picking this up. So, to have a platform like Jama Connect out there to aid in this is going to be very important.
Jama Software: What resources are available to people that you know of to help them comply with the new standards?
Satyajit Ketkar:
Most of the approved, certified, notified bodies in Europe — some of the ones that I mentioned before like BSI, TÜV SÜD, DEKRA — are a great resource. They should have a quick cheat sheet on their website. You can also go online to any of the medical device journals, and they have a pretty good high-level breakdown of EU MDR. It’s more on the regulatory side than a development-process side, but it gives you the breakdown of what articles are necessary, what annexes need to be reviewed, and what order to do it in.







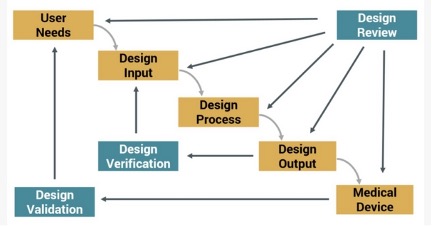



 If you haven’t already, check out
If you haven’t already, check out