Tag Archive for: Requirements & Requirements Management
Tag Archive for: Requirements & Requirements Management
This blog overviews our Datasheet, “Obeo Integrates Capella Models with Jama Connect® Requirements and Tests for Live Traceability™.” Download the entire thing HERE.
OBEO PUBLICATION FOR CAPELLA + JAMA CONNECT GIVES YOU MORE
INCREASE DEVELOPMENT EFFICIENCY: Replace error-prone manual checking of new and changed items with automated updates across the development toolchain and throughout the development lifecycle.
SIMPLIFY VISIBILITY, COLLABORATION, AND REVIEWS: Make it easier for stakeholders to view and interact with requirements and test management data in Jama Connect automatically synchronized with visual models in Capella or published to a server.
MINIMIZE RISKS WITH LIVE TRACEABILITY: Identify issues earlier to minimize defects and rework by ensuring that engineers and other stakeholders can quickly and easily access the latest and most complete information for any
requirement at any time.
Integration Overview
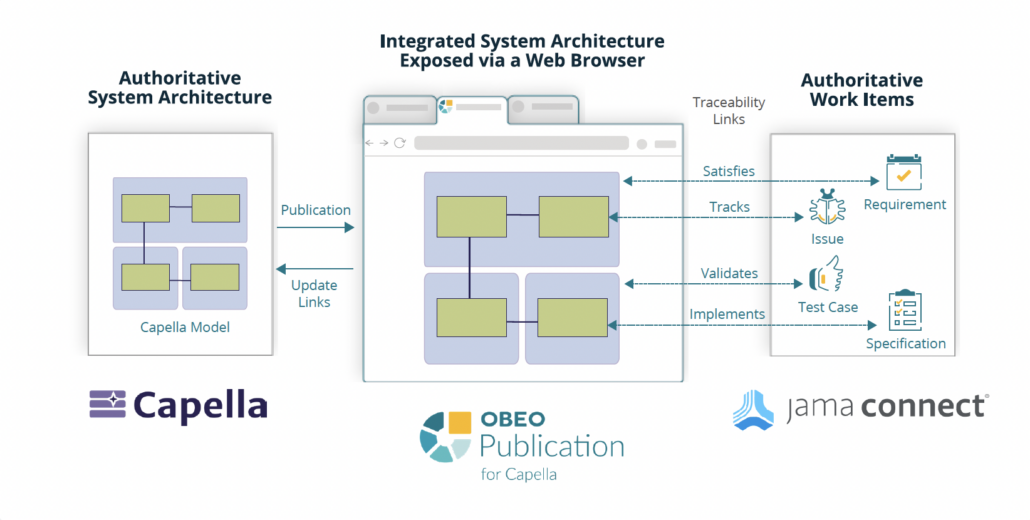
Obeo and Jama Software have partnered to provide a seamless view between Capella’s model-based systems engineering (MBSE) and Jama Connect’s requirements and test management solutions.
Obeo’s Publication for Capella integration platform makes it possible to visualize requirements and tests in Jama Connect and their relationships to Capella elements and diagrams. This integration leverages Jama Connect’s best-in-class API to link requirements to visual models and provide versioning and baselining capabilities for visual models in Capella, Cloud for Capella, and Team for Capella.
By enabling seamless traceability between design models and requirements, organizations gain better visibility into traceability coverage and regulatory compliance. Stakeholder input and reviews are possible by accessing and interacting with models published online, system data without the need to install Capella, which accelerates decision-making and reduces overhead.
Users can quickly and easily create links from Capella elements and their diagrams to Jama Connect requirements and test cases using simple drag-and-drop or copy/paste techniques in Capella. When changes to linked items in Jama Connect occurs, links are automatically synchronized between the Publication for Capella server and Jama Connect, including adding or removing links, to ensure model alignment. When diagrams are added, modified, or removed in Capella, they are automatically published as attachments to linked items in Jama Connect.
When Change Impact Becomes Chaos: A Business Analyst’s Survival Guide
Requirements change. It’s not a possibility. It’s a certainty. Priorities shift mid-sprint, regulators update compliance standards, and stakeholders introduce new dependencies long after a project has gained momentum. For business analysts (BAs), this constant flux creates a ripple effect that’s difficult to track and even harder to control.
The challenge isn’t change itself. The real problem is understanding what that change affects. When a regulatory requirement updates or a business rule shifts, business analysts (BAs) need answers fast: What components are impacted? Who needs to review the changes? What’s already been built, tested, or signed off?
Without a clear picture of these connections, change impact becomes guesswork. Teams scramble to notify the right people, rework spreads across departments, and costly surprises surface late in the development cycle, which is exactly when they’re most expensive to fix.
This post breaks down why fragmented traceability leads to chaos, how automated live traceability transforms the way teams respond to change, and what practical steps BAs can take to regain control.
BA teams often manage requirements through a patchwork of tools that were never designed to work together. Word documents capture business needs. Jira or Azure DevOps track delivery. Excel spreadsheets attempt to maintain traceability. Email threads handle approvals.
Each tool serves a purpose on its own. Together, however, they create a fragmented environment where critical relationships between requirements, design elements, test cases, and deliverables are invisible. When something changes, BAs must dig through documents, cross-reference spreadsheets, and send multiple follow-up messages just to confirm what’s affected.
This process is slow, error-prone, and frustrating for everyone involved. And the problem compounds as project complexity grows.
The Bottleneck Effect on Large Programs
On large transformation programs with multiple stakeholders, fragmented traceability becomes a serious bottleneck. Business, IT, and QA teams often work from different versions of the same document; each believing their source is current. BAs end up playing referee, reconciling conflicting information, chasing approvals, and rebuilding traceability matrices from scratch before every audit.
The downstream consequences are significant. Rework increases. Timelines slip. Defects that could have been caught early surface during user acceptance testing (UAT), when fixes are far more costly to implement. According to research on software development costs, defects identified during UAT can cost up to 15 times more to fix than those caught during the requirements phase.
Where Manual Impact Analysis Breaks Down
Manual impact analysis relies heavily on institutional knowledge to know which requirements connect to which design elements, which test cases cover which features, and which stakeholders own which components. When that knowledge lives in someone’s head rather than a shared system, any staff change or project transition creates gaps.
These gaps surface at the worst possible moments. A critical dependency gets missed during a change review. A test case that covers a recently modified requirement doesn’t get updated. A regulatory change triggers a cascade of downstream updates that no one mapped out in advance. Each of these scenarios is preventable, but only if teams have reliable visibility into the connections that matter.
Live Traceability™ as a Solution
Automated, Live Traceability changes how teams manage change impact at a fundamental level. Rather than manually reconstructing connections between requirements, design elements, test cases, and deliverables, teams can see these relationships in real time and act on them immediately.
When a requirement changes, the impact becomes visible instantly. BAs can identify affected components, notify relevant stakeholders, and assess whether anything downstream needs adjustment before the ripple effect takes hold.
Faster Decisions, Fewer Surprises
Live Traceability accelerates decision-making because the information BAs need is always current and always accessible. There’s no waiting for someone to update a spreadsheet or cross-reference a document. The connections are maintained automatically, so when a change occurs, the system surfaces what’s affected rather than leaving teams to discover it manually.
This visibility helps teams move faster without sacrificing quality. Changes get validated earlier in the development cycle, reducing the likelihood of expensive rework during UAT or post-release. Teams maintain alignment across departments because everyone works from the same system of record, not on parallel versions of a document that diverged weeks ago.
Alignment Across Departments
One of the most significant benefits of live traceability is the reduction of cross-functional friction. When business, IT, and QA teams share a single, authoritative view of requirements and their connections, communication improves dramatically.
BAs spend less time reconciling conflicting information and more time contributing to strategic decisions. Stakeholders get faster answers to change impact questions. Development teams understand exactly which requirements drive which deliverables, reducing ambiguity during implementation. The entire organization benefits from a more reliable, transparent process.
Compliance and Audit Readiness Without the Scramble
For teams operating in regulated industries such as medical devices, automotive, and aerospace and defense, regulatory compliance and audit preparation consumes considerable time and resources. Traceability matrices need to be current, complete, and accurate. When traceability is maintained manually, preparing for an audit means recreating documentation that should have been maintained throughout the project.
Live Traceability eliminates this problem. Because connections between requirements, design, and testing are maintained automatically and continuously, audit-ready documentation is always available. Teams don’t need to scramble because the record is already there.
Recognizing the problem is the first step. Acting on it requires a clear-eyed assessment of where your current process creates friction.
Start by measuring your current impact analysis process. Ask how long it takes your team to complete a change impact assessment today. How many tools and conversations are involved? How often do downstream surprises emerge during testing? How much time goes into rebuilding traceability matrices before audits? These questions surface the true cost of manual traceability, which is often much higher than teams realize.
Identify where the gaps are largest. In most organizations, the weakest link is the connection between requirements and testing. Changes to requirements frequently don’t trigger updates to test cases, leaving gaps that only become visible during UAT. Mapping these gaps helps teams prioritize where automation will deliver the greatest impact.
Evaluate tools designed specifically for requirements management. General-purpose platforms like Jira and Confluence are valuable for project delivery, but they weren’t built to maintain end-to-end traceability. Purpose-built requirements management tools offer automated traceability, change impact analysis, and audit trails that general-purpose platforms can’t replicate. Look for solutions that integrate with your existing delivery tools rather than replacing them. The goal is to close gaps, not add complexity.
Build change impact analysis into your workflow. Even with the right tools in place, process discipline matters. Establish a clear protocol for how change requests trigger impact assessments. Define who owns the review, who needs to be notified, and what criteria determine whether downstream components require updates. Embedding these steps into the standard workflow prevents the informal processes that create gaps.
Invest in team capability. Tools are only as effective as the people using them. Ensure BAs and project teams understand how to use traceability features, how to interpret impact analysis outputs, and how to communicate change implications to stakeholders clearly and quickly.
Taking Back Control of Change Impact
Change will always be part of complex software and systems development. The question every BA team must answer is not how to prevent change. It’s whether your team can respond to it with confidence or scramble to keep up.
Fragmented, manual traceability makes scrambling the default. Automated, live traceability makes confident, rapid response possible. Teams that invest in the right tools and processes gain more than efficiency. They gain the ability to absorb change without chaos by delivering projects that stay on track, meet compliance requirements, and reflect the most current understanding of what stakeholders actually need.
The cost of doing nothing compounds with every missed dependency, every late defect, every audit that requires days of preparation. The cost of acting is a more structured, connected, and resilient way of working that pays dividends across every project that follows.
Note: This article was drafted with the aid of AI. Additional content, edits for accuracy, and industry expertise by Kirsten Moss and Mark Levitt.
IEC 62304 Edition 2: What to Expect and Why It Matters
IEC 62304, the international standard governing medical device software lifecycle processes, is undergoing its first major revision in nearly 20 years. While the upcoming second edition aims to clarify requirements and better reflect modern software practices, it also intentionally preserves the stability manufacturers rely on for compliance.
This webinar, co-presented with Medical Device HQ, provides a clear, practical view into the direction of IEC 62304 Edition 2, directly from someone involved in drafting the standard.
In this session, Christian Kaestner, member of IEC TC 62 / SC 62A and contributor to IEC 62304 Edition 2, joins Tom Rish, Head of GTM Strategy at Jama Software to explain what is changing, what is deliberately not changing, and what these updates mean in practice for medical device and Software as a Medical Device (SaMD) manufacturers.
Key Takeaways:
Understand why IEC 62304 is being revised and the core objectives of Edition 2
Learn how the IEC standardization process has shaped the scope and content of the revision
Discover what the draft edition says about key topics like SaMD and artificial intelligence (AI).
Hear why AI will not be a significant part of IEC 62304 — a deliberate design choice you need to understand.
Find out which proposed changes are most likely to affect your organization and get practical advice on how to prepare.
Get a grounded, standards-focused perspective on IEC 62304 Edition 2 and what it means for your software lifecycle processes.
THE VIDEO BELOW IS A PREVIEW – WATCH THE ENTIRE PRESENTATION HERE
TRANSCRIPT PREVIEW
Christian Kaestner: Thank you, Tom. So to avoid seeming too nerdy, I just want to share more of my human side, because we got a bit technical before, perhaps. I have three adult children, four cats, four beehives, 15 hens, and I love gardening. So now you know everything about me. Back to my nerd side, and what could be better than starting with a disclaimer? This is not an official IC presentation. It’s my perspective on the ongoing work, and I’m one of 73 experts on the project team. Even though I haven’t asked my 70-plus colleagues, I believe today’s presentation is a fair summary of the current status. I would say we are roughly 25 experts actively involved in the work, and it’s definitely a team effort. I just want to emphasize that it’s not my own work. Before we get going, I’m curious, what type of software do you mostly work with? So please participate in the poll and let me know what field of interests you have or what you’re working with.
Let’s see. We’ve got some poll answers. We’ll let you get some time to find the right window. So I see we have a combination, so a mix of both. It’s actually one-third each. Have you all agreed on voting like that? It’s interesting because sometimes there’s a tendency to believe that everything is sampled nowadays, but there’s still quite a big bunch of software in the medical SiMD components out there. Okay. Thanks a lot for participating. Let’s continue. While I’m presenting, perhaps you get bored, but if you do, please share your thoughts in the chat. I’m generally interested in hearing about any challenges you face with the current version of the standard, or what guidance do you seek regarding AI in medical devices? And as Tom mentioned before, please also use the dedicated Q&A box if you have any questions, and I’ll do my best at the end of this webinar to answer to your questions.
Kaestner: Now, let’s get started. Before we discuss the upcoming changes in the second edition, I want to share the reasons behind these changes, because if you don’t understand what’s behind it, it might be difficult to understand why changes are implemented. I also want to provide insight into the behind-the-scenes process involved in developing standards. The first version of IEC 62304 was published in 2006 to meet regulatory requirements and expectations for standardized guidance on software development. In 2015, the standard was amended to include guidance on managing legacy software. The D, with the introduction of the legacy clause, was to help manufacturers bring products developed before the establishment of the standard into compliance. Additionally, there were minor changes to the software safety classification aimed at helping manufacturers manage the classification process more effectively, rather than consistently ending up in class C. Whether that really worked or not, that can of course be debated.
In addition, a major change is the scope shift from medical device software to health software. This adjustment is made to full align with IEC 820304-1, which is the product standard for standalone software, often called SaMD. There will also be simplification of the software safety classification by reducing the number of levels from three to two. And whether that will be simplification or not, we will get back to that, because it comes with some challenges as well. Lastly, considering that the current version is nearly 20 years old, I expect a new addition to include a level of, let’s say, modernization to reflect today’s state of the art. I haven’t seen any updated timeline for the project, but I’m guessing, or perhaps hoping, crossing my fingers, it will be finished by 2028, but you never know. And I’ll come back to why I don’t know, because there are some uncertainties on this path.
Kaestner: For those unfamiliar with the standardization process, here comes a short summary. It begins with a working drop, which is often referred to as a new work item proposal, and that’s especially when it develops the creation of new standards. It’s worth noting that there was a previous attempt to develop a second edition of IEC 62304. Unfortunately, this effort stalled due to challenges in achieving consensus among key stakeholders. However, an important outcome of that previous work was a design specification that now functions as a working draft and leads the project. The specification has guided the development of the first committee draft and will continue to inform the project moving forward. So you can see it’s kind of the rail guards for the project. Although not all national committees are happy about the design specification, it has gathered a majority acceptance, and the project team is keen not to deviate too much from the specification to avoid yet another stopped project.
Here is a summary of the key guiding principles for the project team. So as mentioned earlier, the scope will change to health software. Three levels will change to two. IEC 62304 is a process standard that relies on other standards for product-relevant requirements. This will be emphasized. So, where do you get, for instance, risk management from? And this is not all. Due to the scope change, normative references to medical device standards is not an option longer. The legacy clause has to some extent been used to cheat, unfortunately, I would say. This has resulted in suggesting moving the legacy clause to an informative annex. State of the art expects some level of architectural planning for all levels nowadays. Annexes shall be developed to cover relations to other standards and modern technologies, and also development methodologies such as agile. You will find a link to the design specification in the webinar resource section.
So if you’re interested in the details, please have a look. It’s just like, I think it’s two to three pages long, so it’s not that massive of a document. With the help of the design specification, a CD or committed draft was developed and sent out for commenting at the beginning of last year.
Together with the CD, a change rationale was provided to explain the changes being made, because some changes could seem like kind of unexpected perhaps, and that’s why the change rationale was provided as well to explain these changes. I’ve selected a few key topics addressed in the document. I will also revisit several of them shortly. So the classification of the former software safety classification will be called process rigor level, and the criteria for the determination of the level will also change. There will be new requirements for AI planning, and there will be clarification on supporting items to be controlled. Simply put, you must control whatever items are needed to recreate the software. Whether that is a compiler, test tools, or source code, it’s up to you as a manufacturer to determine what you need to control. The current version has some requirements for communicating with users and regulators. This is typically a product requirement and will be shifted towards the requirement for planning what information shall be communicated to whom and when.
The Simplification of the EU MDR: What MedTech Needs to Know
The European Medical Device Regulation (EU MDR) continues to present challenges for quality and regulatory teams, as well as for the medical device industry more broadly, years after its publication. The updated legislation was introduced with the objective to improve patient safety and harmonize regulatory standards across EU Member States.
Four years after the initial date of application of the regulation, the medical technology industry has gained enough experience to assess its practical impact. While the regulation has strengthened oversight and transparency, it has also introduced complexity, capacity constraints, and unintended consequences that affect product development and market access.
The European Commission has recently proposed a simplification of the EU MDR, supported by a targeted evaluation of both its successes and its limitations. In the following sections, we review the regulation’s background, summarize the key findings of the recent evaluation, and outline practical steps organizations can take to adapt to evolving regulatory expectations.
A Brief History Of The EU MDR
To understand the current regulatory climate, we must look at the system that preceded it. The Medical Device Directives governed the European market since the 1990s. While effective for a time, serious safety events exposed critical flaws in the system. High-profile incidents involving industrial-grade silicone in breast implants and complications with metal-on-metal hip replacements made it clear that the directives lacked sufficient clinical oversight.
The European Union responded by adopting the MDR in 2017, aiming to ensure that only safe, performant devices reached patients. The new framework sought to create a robust, transparent, predictable, and sustainable regulatory environment. It introduced several major changes:
Stricter requirements for clinical evidence and post-market surveillance.
More rigorous criteria for the designation and oversight of notified bodies.
The establishment of the European database on medical devices (EUDAMED) to improve traceability.
The introduction of the Unique Device Identification (UDI) system.
The regulation officially applied in May 2021, but the scale of these changes brought significant growing pains for the industry.
Assessing The Current State Of The EU MDR
The recent European Commission Staff Working Document Evaluation of medical and diagnostic device regulations for the proposal to simplify and lessen regulatory burdens provides a comprehensive look at how the regulation operates today. The findings show a mixed reality. The framework successfully strengthened safety protocols, and vigilance activities increased significantly, giving authorities better tools to detect and manage risks. However, the implementation revealed severe bottlenecks that constrain market functioning.
Innovation And Competitiveness Challenges
The regulation places massive administrative burdens on medical device manufacturers. The assessment highlights that small and medium-sized enterprises are facing disproportionately high compliance costs. Manufacturers reported expenses of approximately €30,000–€250,000 per clinical evaluation, depending on device class and study complexity, with the highest costs incurred for Class III devices. The heavy financial and administrative toll forced many companies to divert resources away from research and development, slowing the pace of innovation. Consequently, some developers began prioritizing market launches in regions with more predictable regulatory pathways, such as the United States.
Notified Body Bottlenecks
Notified body capacity became a major hurdle early in the transition. Lengthy designation processes and extensive documentation requirements led to certification delays lasting 13 to 24 months. These delays threaten the availability of critical medical technologies. To prevent widespread market shortages, the European Commission extended the transition periods to December 2027 for legacy high-risk devices and December 2028 for legacy medium-risk devices.
Transparency And Predictability Gaps
While the regulation aimed to improve legal certainty, stakeholders still struggle with ambiguous definitions and inconsistent applications of the rules. The Medical Device Coordination Group issued over 100 guidance documents, but practical implementation challenges and differences in interpretation persist, making operational clarity harder to achieve in some areas. Furthermore, the slow rollout of EUDAMED delayed the intended transparency goals. Without the fully mandatory database in place, manufacturers continued to grapple with fragmented national registration systems.
The Threat To Orphan Devices
The assessment explicitly noted the threat to niche and orphan devices. Because these products serve small patient populations, they generate lower revenue. The high cost of gathering new clinical data under the MDR led some manufacturers to discontinue these essential devices, posing a direct risk to vulnerable patient groups.
Acknowledging these significant challenges, the European Commission concluded that the regulatory framework requires targeted adjustments. The upcoming simplification revision of the EU MDR, proposed in December 2025, aims to restore European competitiveness and support innovation without sacrificing patient safety.
The simplification effort focuses on reducing unnecessary administrative complexity and creating a more proportionate system. Regulatory bodies plan to streamline reporting obligations and eliminate overlapping assessments. This includes creating tailored requirements for low-risk devices and well-established technologies, which currently face burdens disproportionate to their actual risk profiles.
For niche and orphan devices, the European Commission intends to develop flexible regulatory pathways. These specialized routes will ensure that patients with rare conditions maintain access to life-saving treatments.
Another major component of the simplification is the centralization and harmonization of notified body oversight. By standardizing practices across Member States and moving away from slow, consensus-based decision making, the European Union hopes to improve predictability and reduce certification timelines. The simplification also embraces digital transformation, permanently expanding the use of electronic instructions for use and paving the way for efficient electronic submission systems.
How To Prepare For Upcoming Regulatory Changes
Preparing for regulatory shifts requires a proactive strategy. Quality and regulatory teams should take steps now to ensure a smooth transition when the simplified rules take effect.
Audit your existing product portfolio: Identify devices that might qualify as well-established technologies or orphan devices. These products may soon benefit from streamlined regulatory pathways, saving your team significant time and resources.
Prepare your data for EUDAMED: Although the full system experienced delays, several modules will become mandatory by May 2026. Transitioning from fragmented national databases to a centralized European system requires clean, organized, and highly accurate data.
Establish early dialogue with your notified body: Clear communication helps you understand specific expectations and prevents misinterpretations that cause certification delays.
Streamline your clinical evidence pipelines: Ensure your clinical evaluation reports and post-market surveillance plans are up to date. Robust data remains the foundation of compliance, even under a simplified framework.
Upgrade your internal documentation systems: Manual documentation methods simply cannot keep pace with dynamic regulatory landscapes. Moving away from static documents is essential for maintaining compliance.
Position Your Team For Success With Jama Connect
For teams facing high process complexity and strict regulatory demands, Jama Connect offers a scalable, automated solution. Our platform streamlines traceability, enhances collaboration, and ensures compliance across global markets.
Jama Connect automates traceability and documentation processes, reducing manual effort by up to 50%. By replacing manual spreadsheets with a centralized digital platform, your team can improve risk analysis and maintain a clear line of sight from initial design requirements to final validation. This level of organization proves critical during notified body audits and helps you bring safe, compliant devices to market faster.
Whether you are a startup launching a novel device or an enterprise managing a massive global portfolio, Jama Connect accommodates your growth. The platform integrates seamlessly with your existing tools, allowing your team to focus on strategic innovation rather than administrative overhead. By modernizing your requirements management today, you position your company to navigate the simplification of the EU MDR with total confidence.
Note: This article was drafted with the aid of AI. Additional content, edits for accuracy, and industry expertise by Tom Rish and Victoria Bruno.
Jama Software is always looking for news that would benefit and inform our industry partners. As such, we’ve curated a series of customer and industry spotlight articles that we found insightful. In this blog post, we share an article from AMA, titled “Augmented Intelligence in Medicine” and originally published on October 21, 2025.
Augmented intelligence in medicine
Artificial intelligence vs. augmented intelligence
The AMA House of Delegates uses the term augmented intelligence (AI) as a conceptualization of artificial intelligence that focuses on AI’s assistive role, emphasizing that its design enhances human intelligence rather than replaces it.
AMA policy on AI development, deployment and use
The AMA is committed to ensuring that AI can meet its full potential to advance clinical care and improve clinician well-being. As the number of AI-enabled health care tools continue to grow, it is critical they are designed, developed and deployed in a manner that is ethical, equitable and responsible. The use of AI in health care must be transparent to both physicians and patients.
In addition to medical devices, AI is increasingly used in health care administration or to reduce physician burden, and policy and guidance for both device and non-device use of health care AI is necessary. Recognizing this, the AMA has developed new policy (PDF) that addresses the development, deployment and use of health care AI, with particular emphasis on:
Health care AI oversight
When and what to disclose to advance AI transparency
Generative AI policies and governance
Physician liability for use of AI-enabled technologies
AI data privacy and cybersecurity
Payor use of AI and automated decision-making systems
Physician sentiments on AI
In 2023, the AMA conducted a comprehensive study of over 1,000 physicians’ sentiments towards the use of AI in health care including current use and future motivations for use, key concerns, areas of greatest opportunity and requirements for adoption. Given the rapidly evolving AI landscape across health care, the AMA repeated the study in late 2024 (PDF). The objectives of this research remain:
Capturing the sentiment among practicing physicians regarding the increased usage of AI in health care
Evaluating AI use cases based on their familiarity, relevance, and usefulness
Identifying key resources and areas of need for physicians to consider implementation of AI tools to their practice
Physicians largely remain enthusiastic about the potential of AI in health care, with 68% seeing at least some advantage to the use of AI in their practice, up from 65% in 2023. We also saw use of AI increase from 38% in 2023 to 66% of physicians reporting they use some type of AI tool in practice in 2024.
However, there are still key concerns as physicians continue to explore how these tools will impact their practices. Implementation guidance and research, including clinical evidence, remain critical to helping physicians adopt AI tools.
Physician sentiments study on AI: AMA’s latest study on physician sentiments around the use of AI in heath care: motivations, opportunities, risks and use cases. Read Now (PDF)
AI is playing an increasingly important role at all stages of the medical education continuum, both as a tool for educators and learners and as a subject of study in and of itself. AI has the potential to transform the educational experience as a part of precision education and transform patient care as a part of precision health. Learn more about how AI can impact medical education.
In October 2025, AMA launched the Center for Digital Health and AI to put physicians at the center of shaping, guiding and implementing AI tools and other technologies that are transforming medicine.
AMA welcomes the federal government’s new 2025 action plan on AI and the opportunity to work with the administration to address key areas in shaping AI regulation, policy and implementation. Learn more.
An AMA issue brief (PDF) provides a brief overview of recent state legislative activity and discusses three key AI policy areas for state legislative/regulatory activity: health plan use of AI, transparency and physician liability.
To develop actionable guidance for AI in health care, the AMA reviewed literature on the challenges health care AI poses and reflected on existing guidance. These findings are published in a paper in Journal of Medical Systems:Trustworthy Augmented Intelligence in Health Care.
The current CPT® code set drives communication across health care by enabling the seamless processing and advanced analytics for medical procedures and services.
AMA offers several resources to provide guidance on the updated CPT® code set for classifying various AI applications as well as advisory expertise through the Digital Medicine Payment Advisory Group (DMPAG). DMPAG identifies barriers to digital medicine adoption and proposes comprehensive solutions on coding, payment, coverage and more. Stay up-to-date on the criteria for CPT® codes, access applications and read frequently asked questions.
In this blog, we cover our recent datasheet, “Deliver Innovative Products Faster with Jama Connect for Consumer Electronics Development” – To download this asset, click HERE.
Deliver Innovative Products Faster with Jama Connect for Consumer Electronics Development
Consumer electronics markets are highly competitive, with pressure on companies to deliver new and improved products fast. Falling short of consumer and supply chain expectations relating to product features, performance, or quality can hurt company reputations with buyers and resellers in the marketplace. Failure to satisfy applicable safety and environmental regulations can lead to
product recalls and payments to consumers and government agencies.
To be successful, companies must be able to develop new or improved products quickly by efficiently managing customer, market, and regulatory requirements across product lines with multiple configurations to meet the needs of buyers and regulators around the world.
Jama Connect for Consumer Electronics Development helps companies reduce time to market in delivering innovative, quality, affordable products to avoid recalls and reengineering, and stay ahead of the competition, and meet customer expectations. It supports compliance with IEC 62368 consumer electronics and IEC 60730 home appliance global safety standards.
Guided, Measurable Product Development: Jama Connect’s intelligent engineering management enables significant reduction in re-engineering and product recalls by guiding the user through the end-to-end product development, automatically detecting gaps and risks across the entire engineering data, and automatically measuring the system and process completion.
AI Engineering Automation:Jama Connect Advisor’s™ AI will automate manual, day-to-day engineering tasks so that engineering can focus on innovation and problem-solving.
Digital Co-development Across the Supply Chain: With Jama Connect’s native co-development and engineering collaboration features, internal development and quality teams and external subcontractors and contract manufacturers can jointly define and develop products in the same source of truth in real time.
Apply Intelligence to Product Line Engineering: Efficiently manage requirements and tests shared by related products in libraries selectable based on the relevant configuration.
Streamline Safety and Cybersecurity Risk Analysis, Testing, and Compliance Reporting: Simplify adherence to applicable safety and cybersecurity regulations with pre-built requirements, automated test case generation for traceability, and templates for exporting compliance reports.
Jama Connect for Consumer Electronics Development Out-of-the-Box
Jama Software’s solution includes the following for developing integrated multidisciplinary consumer electronics products, subsystems, and software platforms:
Product, subsystem, and software development items
Product line engineering
Safety, risk, and cybersecurity regulation items
Co-development and digital thread process recommendations
Report templates
Organizations at the forefront of consumer electronics innovation recognize Jama Connect as the tool that provides a competitive advantage in the market by enabling the acceleration of development and bringing innovative products to market faster.
To learn more about managing your consumer electronics and home appliance requirements, tests, and risks more intelligently and efficiently with Jama Connect, visit jamasoftware.com
Purpose-built support for automotive systems and components, ensuring compliance with ISO 26262, ISO 21434, ASPICE, and other critical industry standards.
Centralized platform for managing requirements, tests, risks, and traceability with end-to-end visibility.
Tools for improving productivity, reducing defects, and ensuring seamless traceability to meet safety and security standards.
With Jama Connect, automotive teams can reduce rework, prevent recalls, and bring products to market faster while staying audit-ready.
Plus, leverage pre-configured frameworks and work with Jama Connect consultants to customize the solution to your exact business needs.
Grant Rhodes: Hello, and welcome to the Jama Connect Features in Five series. My name is Grant Rhodes, and I’m a Senior Solutions Consultant here at Jama Software.
Today, we’ll be walking through the automotive solution. Jama Software provides robust tools and solutions to help automotive developers streamline compliance with ISO 26262, ISO 21434, ASPICE, and other critical industry standards.
Centralized Management with Jama Connect
Through Jama Connect, teams can manage requirements, tests, risks, and traceability in a centralized platform, ensuring end-to-end visibility across the development cycle.
This level of traceability is crucial for demonstrating adherence to safety and security standards. It enables significant productivity and quality improvements, dramatically reduces the risk of product delays, cost overruns, defects, rework, and recalls, and ultimately results in faster time to market.
The Jama Connect automotive solution is a complete set of frameworks, example projects, and procedural documentation intended to accelerate the implementation of Jama Connect for organizations developing automotive systems and components. The foundation of our automotive solution is Jama Connect, our industry-leading, best-in-breed requirements management solution.
Purpose-Built for Automotive Development
Purpose-built to track the requirements of complex systems and reduce risk and inefficiencies of document-based legacy systems, the automotive solution allows teams to start working in Jama Connect with zero setup and configuration time. Alternatively, teams can work with a Jama Connect consultant to customize the solution to meet their company’s exact business needs.
Rhodes: The automotive solution frameworks come in the form of projects in Jama Connect. These include definitions of item types and relationships as well as example project hierarchies that are aligned to key industry regulations. Here, I have the base automotive framework project open.
This image represents the traceability information model applied within this project and visualizes the allowable data types and relationship rules. Requirements are covered by lower-level requirements or design elements and are related to test case items to prove compliance. In addition to the item types and relationships, each framework also contains a project structure designed to highlight important sets of data.
These models and hierarchies have been designed using input from industry best practices and are usable out of the box. However, Jama Connect is configurable and can be customized to meet any needs specific to your organization or product development cycle.
Sample Projects for Hands-On Experience
In the project tree, we can see that in addition to the framework projects, the solution also delivers sample projects.
These utilize a framework but are populated with sample data. In these projects, teams can get hands-on experience with the defined item types and relationships. For example, opening the automotive safety and security sample set project, we can see an example of a hazard analysis and risk assessment (HARA).
Rhodes: The automotive solution also includes many export templates and reports for generating HTML, PDF, Word, and Excel outputs from the system.
Some reports are generic and included in all Jama Connect instances. Others are targeted for automotive customers, providing content and formatting specific to industry needs. For example, the HARA and TARA reports give the ability to export safety and security items from a project. Since the sample set project is populated with data, we can use it to better understand the outputs that these reports deliver.
Conclusion
Thank you for watching this Features in Five session on the automotive solution for Jama Connect. Existing customers, if you want to learn more, please reach out to your Customer Success Manager or Consultant. New customers, if you are not yet a client, please visit our website at JamaSoftware.com to learn more about the platform and how we can help you optimize your development process.
A Practical Guide to Translating User Needs into Design Inputs
As a former product development engineer, I remember the pressure to start designing immediately. I’d jump straight into CAD models and prototypes, eager to build the next innovative medical device. But sometimes, this meant I overlooked a critical first step: truly understanding what the end-user needed. This often led to features that missed the mark and created a mountain of documentation rework to justify our design choices after the fact.
Many engineers in the medical device space get stuck in this cycle. They struggle to distinguish between user needs and design inputs, or they don’t know how to translate a general user request into a measurable engineering requirement. This confusion isn’t just inefficient; it’s a compliance risk that can delay projects and frustrate teams who would rather be designing and testing than drowning in paperwork.
TL;DR: User needs are high-level goals describing what a user wants a device to do, while design inputs are the specific, measurable engineering requirements needed to meet those needs. Following a structured process to translate user needs into traceable design inputs is essential for complying with FDA regulations and building products that succeed.
What are User Needs? The Foundation of Your Design Control Process
User needs are the starting point for the entire medical device design control process. They are high-level, qualitative statements that capture the goals and expectations of the device’s intended users. Think of them as the “what” from the user’s perspective.
These needs are derived from various stakeholder needs, which can include patients, surgeons, nurses, technicians, or even hospital administrators. The key is to capture their desired outcomes without dictating a specific technical solution.
According to FDA 21 CFR 820.30, the design control process begins with establishing and maintaining procedures to ensure that the design requirements are appropriate and address the intended use of the device, including the needs of the user and patient.
Examples of User Needs:
A surgeon needs the device to provide clear visualization in a smoke-filled environment.
A home-care patient needs the device to be simple to operate without assistance.
A nurse needs the device to be easily and quickly sterilized between uses.
What are Design Inputs? The Blueprint for Your Device
If user needs are the “what,” design inputs are the “how”— from an engineering perspective. They are the detailed, objective, and verifiable requirements that describe the performance, physical, and functional characteristics of the device. Every design input must be traceable back to a specific user need.
These inputs form the technical blueprint that guides the entire development process. They must be unambiguous and measurable so that you can later prove the device meets them through design verification activities.
Key takeaway: Without clear design inputs, you have no objective criteria to design against or to test your final product.
Examples of Design Inputs (translated from the user needs above):
User Need: A surgeon needs clear visualization.
Design Input: The device’s camera shall operate in temperatures up to 60°C.
Design Input: The device’s lens shall be coated with an anti-fog agent.
User Need: A patient needs a simple device.
Design Input: The device shall have no more than three buttons for all primary functions.
Design Input: The device startup sequence shall complete in under 5 seconds.
User Need: A nurse needs easy sterilization.
Design Input: The device housing shall be made of medical-grade stainless steel 316L.
Design Input: The device shall withstand at least 100 autoclave sterilization cycles at 134°C.
A 4-Step Guide for Translating User Needs to Design Inputs
Translating vague user needs into concrete design inputs is a skill. It requires a systematic approach to ensure nothing is lost in translation. Following these steps will help you create a robust foundation for your medical device design control process.
Step 1: Gather and Define Clear User Needs
Before you can translate needs, you must capture them accurately. This involves engaging directly with your stakeholders through methods like interviews, surveys, and observational studies. Focus on understanding their goals and pain points. Write the user need from their perspective, avoiding technical jargon.
Step 2: Deconstruct Each User Need
A single user need can contain multiple implied requirements. Break down broad statements into their core components. For a need like, “The device must be portable,” ask clarifying questions:
What does “portable” mean to the user? Carried in a pocket, in a bag, or on a cart?
How long does it need to operate without being plugged in?
In what environments will it be used?
Step 3: Write Quantifiable and Verifiable Design Inputs
This is the most critical step. Convert each component of the user need into a specific, measurable requirement. A good design input is unambiguous and testable.
Use “shall” statements: This is standard practice for writing formal requirements.
Be specific: Instead of “lightweight,” write “The device shall weigh less than 500 grams.”
Make it measurable: Instead of “a long battery life,” write “The device shall operate continuously for a minimum of 8 hours on a single charge.”
Step 4: Establish and Maintain Traceability
Every design input you create must be linked directly back to the user need it helps fulfill. This traceability is not optional; it’s a regulatory requirement and the backbone of your medical device file. This link proves that your design is directly driven by user needs and that every requirement has a purpose.
Streamline Your Design Control Process with the Right Tools
Managing the complex web of user needs, design inputs, risks, and verification activities in spreadsheets or documents is a recipe for errors and audit findings. This is where modern requirements management platforms can transform your workflow.
Live Traceability™: Automatically create and visualize the links between user needs, design inputs, test cases, and other artifacts. This ensures you are always audit-ready and can easily analyze the impact of any changes.
Reuse and Libraries: Stop reinventing the wheel. Create libraries of common requirements, like those for specific standards or product lines, and reuse them across projects to ensure consistency and save valuable time.
AI-Powered Insights: With Jama Connect Advisor™, you can leverage AI to analyze your requirements for quality. Get instant feedback on whether your design inputs are clear, complete, and verifiable, helping your team write better requirements faster.
Q: Can a single user need lead to multiple design inputs? A: Yes, absolutely. A high-level user need like “the device must be safe for clinical use” will be broken down into dozens or even hundreds of specific design inputs related to biocompatible materials, electrical safety standards, alarm functionalities, and much more.
Q: What’s the difference between design inputs and design specifications? A: This is a common point of confusion. Design inputs define what the device must do (the requirements). Design specifications (also known as design outputs) describe how the device will meet those requirements. They are the tangible results of the design process, such as drawings, material specifications, and source code. People often think of the design outputs as the “recipe” showing how to build the device.
Q: How do I handle conflicting user needs? A: It’s common for different stakeholders to have competing needs (e.g., a large screen for visibility vs. a small size for portability). This requires a structured process of prioritization, risk analysis, and trade-off discussions with the project team and stakeholders. The key is to document these decisions and the rationale behind them within your design history file.
Master Your Design Inputs and Accelerate Innovation
Bridging the gap between user needs and design inputs doesn’t have to be a source of frustration. By adopting a structured process and leveraging the right tools, you can eliminate ambiguity, ensure compliance, and free up your engineers to do what they do best: build innovative products that improve lives.
BrightInsight Drives Efficiency Using Jama Connect
ABOUT BRIGHTINSIGHT
BrightInsight is a trusted partner for top life sciences companies, delivering regulated digital health solutions like Software as a Medical Device (SaMD) that transform patient care. In collaboration with leading BioPharma and MedTech firms, BrightInsight has developed pioneering solutions across more than 11 therapy areas.
CUSTOMER STORY OVERVIEW
Operating in a highly regulated industry, BrightInsight navigates strict deadlines, complex regulatory requirements, and dynamic client expectations. To maintain its competitive edge, the company needed a robust requirements management tool to refine its processes. A key challenge was integrating cybersecurity into its overall patient safety risk management framework.
By implementing Jama Connect, BrightInsight created a novel, integrated approach to risk management. This allowed them to automate traceability, optimize documentation, and significantly improve project efficiency. As a result, BrightInsight has reduced risk assessment activities by more than 50% and accelerated project timelines, all while ensuring comprehensive cybersecurity and patient safety.
“We’ve been able to link cybersecurity risks back into patient hazards, which is something many companies struggle to do. Jama Connect’s traceability and item relationships make that possible.” – Lucas Holt, Director of Systems Engineering, BrightInsight
WITH JAMA CONNECT, USERS EXPERIENCE:
Ability to seamlessly link cybersecurity risks to patient safety hazards, creating comprehensive risk models that improve safety and compliance.
Optimized workflows and reusable requirement repositories help users reduce project timelines by months, expediting product launches and increasing efficiency.
Real-time feedback tools and robust traceability features streamline collaboration while ensuring audit-ready documentation that impresses regulators.
“Jama Connect really forces you to think about traceability right from the get-go of the project, where in the old way of doing it, traceability was often an afterthought. It’s the right way to do it.” – Lucas Holt, Director of Systems Engineering, BrightInsight
CHALLENGES
Before implementing Jama Connect, BrightInsight faced several industry-wide challenges that hindered its ability to innovate safely and efficiently.
Fragmented Risk Management: Managing cybersecurity and patient safety risks in separate streams resulted in disjointed assessments. This separation made it difficult to understand the true impact of a cyber threat on patient health.
Inefficient Documentation: Manual processes using Word and Excel were inefficient, prone to error, and limited collaboration across global teams.
Cumbersome Traceability: The document-first approach made tracing relationships between requirements, risks, and test cases a time-consuming manual task, often delaying regulatory submissions and product launches.
BrightInsight leveraged the powerful features of Jama Connect to build a unified and efficient development process, with a groundbreaking approach to risk management.
A Novel Approach to Cybersecurity Risk The most significant change was integrating cybersecurity directly into patient safety risk management. Using the powerful traceability features in Jama Connect, BrightInsight linked cybersecurity threat models directly to patient hazards. This innovative structure allows them to create comprehensive risk models connecting assets, vulnerabilities, and threats to specific patient safety outcomes.
Item-Based Management for Seamless Traceability Jama Connect’s item-first approach enabled BrightInsight to manage traceability from the very beginning of a project.
Optimized Reuse and Standardization BrightInsight created reusable requirement repositories, particularly for common elements like cloud-based services. New projects can now pull from this “off-the-shelf” library of pre-built modules with fully linked requirements, risks, and testing frameworks.
Streamlined Reviews and Collaboration Jama Connect’s Review Center replaced lengthy meetings with real-time, asynchronous collaboration. Teams can now access shared projects, provide feedback, and monitor revisions, dramatically improving review timelines.
“The reusability of items, requirement sets, and risk management really shines for us, it has significantly reduced our design cycles.” – Lucas Holt, Director of Systems Engineering, BrightInsight
OUTCOMES
Since implementing Jama Connect, BrightInsight has achieved measurable improvements, solidifying its position as an industry leader.
Faster Cybersecurity Risk Assessments: By integrating and standardizing its processes, the team reduced the time for cybersecurity and patient safety risk evaluations from six weeks down to just two.
Boosted Overall Efficiency: Optimized workflows have expedited product launches, allowing BrightInsight to reduce project timelines by three to six months for its pharma clients.
Enhanced Regulatory Confidence: During audits, BrightInsight can now produce audit-ready documentation with full traceability instantly. This organized, structured approach leaves auditors impressed and confident in their compliance.
Improved Client Satisfaction: Faster turnarounds and greater assurance of quality documentation have led to increased trust and stronger, more successful business relationships.
Preventing a $400 Million Mistake: Why Digital Traceability is Critical in AEC
On November 19, 2025, Amazon announced the indefinite closure of its LIT1 fulfillment center — a $400 million hub at the Port of Little Rock — eliminating 4,100 planned jobs. The reason cited? “Unfixable flaws” in the structure (see article). It turns out that the company’s largest fulfillment center in Arkansas that has been operating since 2021 wasn’t designed to be compliant with the Arkansas Fire Prevention Code that includes seismic safety provisions relevant to the New Madrid Seismic Zone that includes Arkansas.
In large-scale construction and engineering projects, disjointed data often leads to failure. When requirements, such as state building codes, safety regulations, or load-bearing specifications, live in static documents or spreadsheets, they become disconnected from the actual design and verification processes.
If state mandated structural requirements conflict with an architectural design, manual processes often fail to flag the issue immediately. By the time the error is discovered, the structure is built, and the flaw may be deemed “unfixable.”
Jama Connect mitigates this risk through Live Traceability™ that creates a digital thread connecting regulatory and other requirements directly to specific design elements and verification tests across the project software toolchain. If a requirement changes or a test fails, the impact is immediately visible across the entire project. This allows teams to identify non-compliance issues in the digital design phase — long before they become physical, expensive, “unfixable” problems.
Building codes are not suggestions; they are mandates that involve complex layers of fire safety, seismic durability, and occupancy standards. Engineering designs used to construct buildings successfully used in some states may not be reusable as is in other states. As one of seven states in the New Madrid Seismic Zone, Arkansas mandates that structures be designed to adequately resist seismic forces during earthquakes.
Using a requirements management platform allows teams to:
Decompose Regulations: Break down the state’s specific complex safety, fire and other building codes into individual, trackable requirements.
Link Verification to Requirements: Ensure every single code requirement has a specific test or verification method linked to it.
Monitor Compliance Status: View real-time dashboards showing exactly which parts of the build are compliant and which are at risk.
In the case of Amazon’s Arkansas hub, a digital engineering compliance framework could have flagged the non-compliant design elements during the building design stage, prompting a relatively quick fix with a cost measured in hours of engineering time, rather than closer of an occupied, active building surrounded by uncertainty about the possibility and cost of bringing the building into compliance.
Reducing Risk Through Collaboration
Complex projects involve diverse teams — architects, structural engineers, regulatory bodies, contractors, and owners. When these stakeholders operate in silos, critical information falls through the cracks.
Jama Connect serves as a single source of truth providing everyone with access to the latest information and enabling collaboration that is crucial for risk management. When a regulatory requirement lacks verification, the lack of traceability is called out. When an engineer proposes a change, the platform analyzes the downstream impact: Will this change violate a safety code? Will it conflict with a mechanical system requirement?
By centralizing communication around the requirements themselves, organizations ensure that everyone is building toward the same, compliant outcome. This alignment is the primary defense against the late-stage discovery of structural failures.
The elimination of 4,100 jobs and the abandonment of a major fulfillment hub underscore a critical industry lesson: manual documentation methods that are prone to human error are insufficient for modern, high-stakes engineering.
To avoid a similar fate, organizations must embrace digital engineering tools that automate traceability and compliance checks so that their teams can reduce manual effort and focus on innovation and safety. Using Jama Connect will help ensure that by the time construction begins, a successful outcome is guaranteed. In an industry where mistakes cost hundreds of millions, digital traceability is the most effective insurance policy available.
Note: This article was drafted with the aid of AI. Additional content, edits for accuracy, and industry expertise by Joe Gould, Kevin Andrewjeski, and Mark Levitt.