Design Transfer: Best Practices for Translating Your Device Design into Manufacturing Specifications
Manufacturing specifications are successful when they result in your medical device being produced consistently, over and over, and meet requirements and expectations, including those of design intent and quality specifications. In this blog post, I’ll share the best practices I follow when translating a medical device design into manufacturing specifications, specifically the drawings and specifications of parts and assemblies.
1: Start early
When thinking about the typical medical device product development paradigm, design transfer is usually depicted as one of the steps right before commercial production. Sometimes it is shown in parallel to design validation, and always after detailed design and engineering and design verification.
However, in reality, design transfer, especially the translation of your device design into manufacturing specifications, should start earlier.
Creating the right manufacturing specifications starts in the detailed design and engineering phase of product development. Manufacturing specifications are the drawings, part specifications, work instructions for assembly, testing requirements, and other elements of the Device Master Record (DMR), or the ‘recipe’ as I like to call it, to be able to produce your product. Thus, it’s key to be already deciding which technologies will be used to produce your parts and using a Design for Manufacturability (DFM) process to make assembly easier and more efficient and reflect those decisions in your drawings and specifications. And like the product development process itself, translating your device design is iterative in nature. Starting in the detailed design and engineering phase allows you to follow the steps further defined below.
2: Involve your manufacturing partners
This is the time to involve your manufacturing vendors for their input as to how to specify the custom parts and assemblies they will be supplying. They are the most knowledgeable regarding the tolerances and limitations their processes can generally provide while you, as the final device manufacturer, are the expert on the final design intent and criticality of the parts or assemblies they are providing. Working together will result in drawings and specifications that can be manufacturable and meet design intent. This is also another good time to incorporate other Design for Manufacturability (DFM) aspects for the largest positive impact to the design.
RELATED: Five Key Design Control Practices that Improve Compliance and Help Develop Better Products
3: Incorporate risk
Translating your design specifications into manufacturing specifications is not a time to omit incorporating risk. Reference your risk analysis to determine which parts have functional and safety implications and tailor the specifications, including the quality specifications accordingly.
4: Remember the quality specifications
Speaking of quality specifications, having a strong quality control plan, based on risk, is also part of a successful translation of design specifications into manufacturing specifications.
Elements of a quality control plan include quality agreements with vendors; part validation expectations; various inspection requirements, including during receipt of first articles, incoming, in-process, and final acceptance testing; and ongoing process monitoring. Aligning expectations with suppliers of critical components is key. I see these quality specifications as partners to the part and assembly drawings and specifications.
Inspection testing is a good example to see the relationship from a design input specification to a manufacturing specification, specifically in this case, to an in-process testing specification. Say your device has a design input specification for a minimum flow efficiency of 80%. The 80% is based on the clinical need of the device. During design verification, the design is tested with a statistically relevant sample size to have an efficiency of 95% ± 2%. This range is also measured during process validation. Thus, in-process testing can be set at an 89% minimum. Note, this is intentionally tighter than the original design input, so that process issues and drift can be detected earlier.
Another example is related to sterilization. Say the design input is for a sterile product to have a one-year shelf life, i.e., the sterile barrier must maintain its integrity for one year. In this case, a Tyvek heat-sealed pouch is selected as the sterile barrier. During detailed design and development, the specific pouch is selected, the parameters for sealing are determined, and the corresponding peel strength of the seal is measured. Then design verification testing verifies that the pouch, when sealed under the selected parameters, does indeed maintain its sterile barrier after one year (accelerated aging). At this point, the corresponding peel strength of the seal can be used as an in-process specification to monitor the sealing performance. This minimum force specification is based on the performance data measured to date (typically from process validation), with adjustments to accommodate the observed manufacturing variability. Thus, the design input to have a one-year shelf life of the sterile product is translated into a manufacturing, in-process testing specification of a minimum peel test force of the seal.
RELATED: Complying with FDA Design Control Requirements Using Requirements Management Principles
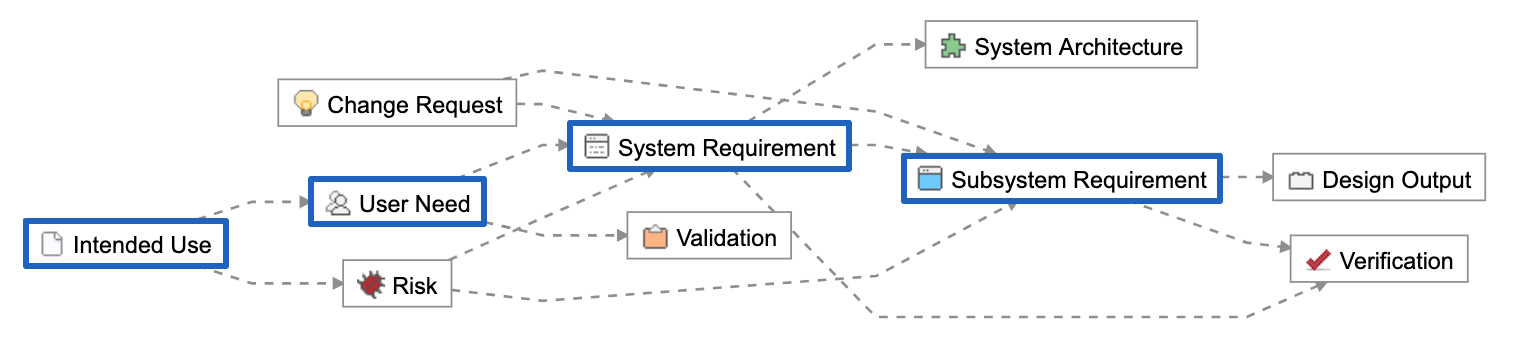
5: Create your traceability matrix
The FDA and other regulatory agencies expect a trace matrix associated with your medical device to show linkages from User Needs through Design Validation.
In the trace matrix, it’s where you can see the direct linkages between part and assembly (design outputs) specifications to corresponding design input specifications. Note that it need not be a 1:1 relationship. Multiple part/assembly specifications can be linked to a design input, and one part/assembly specification could fulfill multiple design inputs.
Here’s a snippet of a trace matrix for a fictional home-use thermometer. Especially for more complex medical technologies, a requirements tool such as Jama Connect® makes it more efficient to create and manage a product’s traceability matrix and ensure there are no gaps. As the Design Output column are manufacturing specifications, this is where you can see the traceability between the design input specifications to manufacturing specifications. It’s in this column where the manufacturing testing requirements in the examples above would be listed.
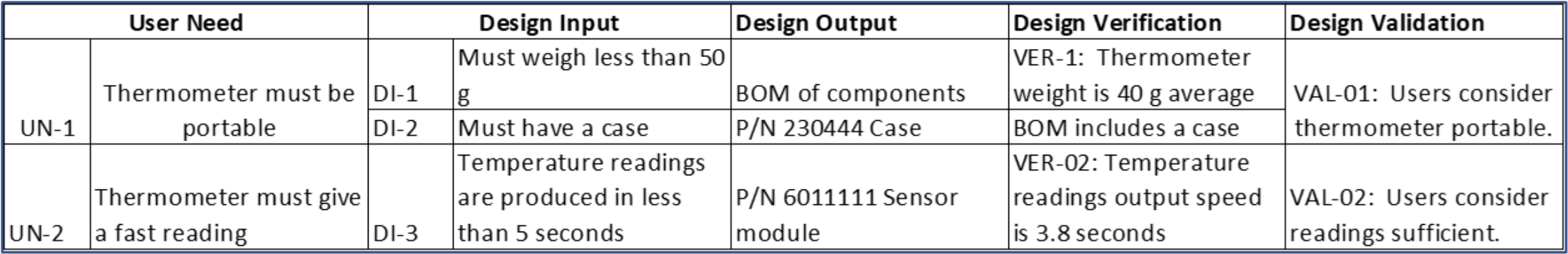
And a well-specified trace matrix is a good tool to use to understand the impact of future design changes, both for changes that occur after the design has transferred to manufacturing, as well as for any changes that may result from design verification and design validation. At times, if the risk is appropriate, you can choose to perform some design verification activities and design validation activities in parallel. Test units for design validation must be production equivalent, thus being able to trace which revisions of the design and resulting manufacturing specifications used units for design verification activities and design validation activities is important and to be able to justify the impact of any differences.
There are many activities to consider as part of manufacturing transfer. These best practices focus on translating the device design into part and assembly drawings and specifications, including the quality specifications, that are a part of ensuring your device is made the right way every time.
RELATED